SAAVpedia
Identification, functional annotation, and retrieval of single amino-acid variants for proteogenomic interpretation
Manual
Second edition 28 Nov 2018
The SAAVpedia is a platform to identify, annotate, and retrieve pathogenic SAAV candidates from proteomic and genomic data. The platform consists of four modules: SAAVidentifier, SAAVannotator, SNV/SAAVretriever, and SAAVvisualizer. The SAAVidentifier provides a reference database containing 5,949,033 SAAVs in 42,134 protein isoforms. The SAAVannotator provides 24 elements belonging to four categories: i) genomic variant, ii) gene/transcript/protein, iii) biological, and iv) clinical information. It provides functional annotation for interpreting condition-specific SAAVs. The SNV/SAAVretriever module enables bidirectional navigation between relevant condition-specific SAAVs and nsSNVs with diverse genomic and proteomic experimental data. Finally, SAAVvisualizer provides various statistical plots based on functional elements of detected SAAVs. Through the proteogenomic interpretation pipeline in SAAVpedia, we discovered disease- or drug-related genes that have SAAVs from breast cancer and glioblastoma using protein-protein interaction network analyses.
4. Usage
– 4.1 Test data
— 4.1.1 Input file for SAAVidentifier
— 4.1.2 Input files for SAAVannotator and SAAVvisualizer
— 4.1.3 Input file for SNVretriever
— 4.1.4 Input file for SAAVretriever
– 4.2 Proteogenomic interpretation pipeline
– 4.3 Web interface
— 4.3.1 SAAVidentifier module
— 4.3.2 SAAVannotator module
— 4.3.3 SNVretriever module
— 4.3.4 SAAVretriever module
— 4.3.5 SAAVvisualizer module
– 4.4 Python stand-alone program
— 4.4.1 Installation of the SAAVpedia Python package
— 4.4.2 Use of the Proteogenomic Interpretation Pipeline
– 4.5 REST API
5. User manual
– 5.1 Web interface panel
— 5.1.1 SAAV sequence database download
— 5.1.2 SAAVidentifier
— 5.1.3 SAAVannotator
— 5.1.4 SNV/SAAVretriever
— 5.1.5 SAAVvisualizer
– 5.2 Python stand-alone program
— 5.2.1 SAAVidentifier.py
— 5.2.2 SAAVannotator.py
— 5.2.3 SNVretriever.py
— 5.2.4 SAAVretriever.py
– 5.3 REST API
— 5.3.1 SAAVidentifier
— 5.3.2 SAAVannotator
— 5.3.3 SNVretriever
— 5.3.4 SAAVretriever
1. Introduction
1.1 Overview
The SAAVpedia is a comprehensive proteogenomic interpretation platform that identifies, annotates, and retrieves SAAVs from proteomic and genomic sequence data to discover true pathogenic variant candidates. SAAVpedia provides a proteogenomic analysis pipeline to reduce and interpret SAAVs. The pipeline contains a database for proteome searches, a SAAV quality checker, a query builder based on clinical, biological, and pharmacological information, and a retrieving and interpreting system (Figure 1-A). The platform consists of four analysis modules provided by the SAAVpedia web, stand-alone, and REST applications (Figure 1-B). First, the SAAVidentifier module automatically performs simultaneous detection, quality control, and assessment of massive amounts of SAAVs from peptide sequencing data. The results are provided by the SAAV-calling format (SCF) that we designed to facilitate information transmission, by which the text file format contains meta-information including the amino acid sequence and protein and genomic position of each SAAV. Second, the SAAVannotator module performs pharmacological, clinical, and biological annotation based on the genomic and proteomic location of identified SAAVs, e.g., annotating phenotype-variant relationships, post-translational modifications (PTMs), and various kinds of variant or gene database identifiers (IDs) such as dbSNP and Ensembl. The SNVretriever module searches for SAAVs that are in the same location as input nsSNV genomic variants and provides the variant list along with diverse information including diseases and drugs. Otherwise, the SAAVretriever module enables querying and navigating SAAVs that were identified from different proteomic data of various phenotype conditions. SAAVpedia is a comprehensive proteogenomic interpretation platform for discovering true pathogenic variant candidates by analyzing genomic and proteomic data. More information is provided on the SAAVpedia web application (https://www.saavpedia.org/)..

Figure 1. SAAVpedia platform for identifying, annotating, and retrieving pathogenic SAAVs from proteomic and genomic data.
1.2 Citation
SooYoun Lee*, Heeyoun Hwang*, Young-Mook Kang, Ji Eun Jeong, Jin Young Kim, Jong Shin Yoo. SAAVpedia: identification, functional annotation, and retrieval of single amino acid variants for proteogenomic interpretation (under review).
1.3 Get Assistance
For assistance, please email <ymkang@kbsi.re.kr> or <heeyounh@kbsi.re.kr>.
2. Data Source
The SAAVpedia sequence database consists of customized neXtProt (https://www.nextprot.org/) and GENCODE (https://www.gencodegenes.org/) databases. The SAAV peptide sequences were tryptic-digested in silico with their altered amino acid sequence using the variant information from peff files of all chromosomes from the neXtProt database, and concatenated in each corresponding protein entry (21). To minimize the misidentification of SAAV peptides and to consider peptides to spectra match alternative splicing variants, we used a 3-frame translated database from GENCODE transcript sequences of protein coding genes (21). These two variant sequences and protein reference sequences from neXtProt were concatenated in a SAAVpedia sequence database. A total of 925 LC-MS/MS files from the CPTAC breast cancer study was downloaded from the CPTAC-DATA portal (https://cptac-data-portal.georgetown.edu/cptac/s/S029).
3. Preparation of input files
3.1 SAAV peptide search
Using the next-generation proteomic pipeline (nextPP), including RawConverter (v.1.1.0.18), Integrated Proteomic Pipeline (v.4.1.2.4), and ProteinInferencer (v.1.0) from the Scripps Research Institute, La Jolla, CA, USA, the SAAV peptides could be searched with < 0.1% and < 1% FDR at the peptide and protein levels, respectively (21). After the proteomic search against the SAAVpedia reference database, SAAV peptides were selected and used in the next step. The DTASelect-filter.txt file generated from nextPP is used to calculate the quality score in SAAVpedia.
3.2 Input file format
3.2.1 SAAV peptide list
The SAAV peptide list is a text-based file of SAAV peptides that contain the amino acid variant or are obtained from in silico tryptic digestion of the protein sequence with the amino acid variant.

3.2.2 SAAV-calling format
The SAAV-calling format (SCF) was designed to facilitate information transmission when the text file format contains meta-information (Table 1-2). SAAVannotator, SNV/SAAVretriever, and SAAVvisualizer accept SCF as the input file. The file contains meta-information lines, a header line, and data lines containing information about a SAAV.

##fileformat=SCFv1.0
##fileDate=20180529
##SAAVpedia sequence database=neXtprot Jan 2017
##Meta information
##SNV_OID=<Description = “EFO ontology ID(OID) of SNV_Phenotype.”, Reference database=”EFO”,Class=”Clinical information”>
##SAAVPedia_ID=<Description = “Internal identifier of SAAV”, Reference database=”SAAVPedia”,Class=”SAAV”>
##SAAV_QS=<Description = “Quality Score(QS),It was calculated quality scoring method and peptide calling score from SAAVpedia and proteomic analysis tools such as IP2″, Reference database=”SAAVPedia”,Class=”SAAV”>
##SAAV_Pvalue=<Description = “P-value from the quality score distribution in SAAV list of input sample”, Reference database=”SAAVPedia”,Class=”SAAV”>
##SAAV_filter=<Description = “filter status,PASS if this SAAV has less than 0.05 SA_Pval. If not, then FAIL”, Reference database=”SAAVPedia”,Class=”SAAV”>
##SAAV_Pos=<Description = “Single amino-acid variant position(Pos),The Proteome reference position of SAAV”, Reference database=”NextProt”,Class=”SAAV”>
##SAAV_Ref=<Description = “Single amino-acid variant reference(Ref) sequence”, Reference database=”NextProt”,Class=”SAAV”>
##SAAV_Alt=<Description = “Single amino-acid variant alteration(Alt) base”, Reference database=”NextProt”,Class=”SAAV”>
##SAAV_RS=<Description = “Peptide reference sequence(RS) which contains one or more SAAVs”, Reference database=”NextProt”,Class=”SAAV”>
##SAAV_AS=<Description = “Peptide alteration sequence(AS) which contains one or more SAAVs”, Reference database=”NextProt”,Class=”SAAV”>
##SAAV_SDM=<Description = “Strand direction matching(SDM) information of SNV and SAAV,If the information is correct, the score is 1 otherwise 0″, Reference database=”NextProt”,Class=”SAAV”>
##SNV_Chr=<Description = “Chromosome(Chr),An identifier from the reference genome”, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_Pos=<Description = “Single nucleotide variant position(Pos),The Genome reference position, with the 1st base having position 1, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_Ref=<Description = “Single nucleotide variant reference(Ref) base,Each base must be one of A,C,G,T,N (case insensitive)”, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_Alt=<Description = “Single nucleotide variant alteration(Alt) base,Comma separated list of alternate non-reference alleles in genome”, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_Strand=<Description = “Genomic variation reference ID (dbSNP, Cosmic)”, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_ID=<Description = “Chromosome(Chr),An identifier from the reference genome”, Reference database=”dbSNP Cosmic”,Class=”SNV”>
##SNV_1000G_T_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_EAS_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome East Asian population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_AMR_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome American population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_EUR_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome European population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_AFR_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome African population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_SAS_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome South Asian population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_1000G_SAS_MAF=<Description = “Minor allele frequency(MAF) in the 1000Genome South Asian population”, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_VT=<Description = “Variant Type(VT),Rare: SNV_1000G_T_MAF <= 0.005,Common: SNV_1000G_T_MAF > 0.05″, Reference database=”1000 Genome”,Class=”SNV”>
##SNV_ESP_OC=<Description = “Occurring(OC) in the Exome Sequencing Project(ESP) variant list,It has two kinds of values(O,X)”, Reference database=”Ensembl variation database”,Class=”SNV”>
##SNV_ESP_AF_MAF=<Description = “Minor allele frequency(MAF) in the ESP African(AF) population”, Reference database=”Ensembl variation database”,Class=”SNV”>
##SNV_ESP_EU_MAF=<Description = “Minor allele frequency(MAF) in the ESP European(EU) population”, Reference database=”Ensembl variation database”,Class=”SNV”>
##SNV_ExAC_OC=<Description = “Occurring(OC) in the Exome Aggregation Consortium(ExAC) variant list,It has two kinds of values(O,X)”, Reference database=”Ensembl variation database”,Class=”SNV”>
##SNV_Phenotype=<Description = “Phenotype term,The information extracted from genomic variation- phenotype relationship database such as Clinvar”, Reference database=”Ensembl variation database”,Class=”Clinical information”>
##SNV_Source=<Description = “Reference database of SNV_Phenotype”, Reference database=”Ensembl variation database”,Class=”Clinical information”>
##SNV_OID=<Description = “EFO ontology ID(OID) of SNV_Phenotype”, Reference database=”EFO”,Class=”Clinical information”>
##SNV_Phe_CLS=<Description = “EFO phenotype class(CLS) name of SNV_Phenotype”, Reference database=”EFO”,Class=”Clinical information”>
##SNV_DB=<Description = “Drug bank(DB) ID,We integrated based on drug target gene that contained one or more SAAVs”, Reference database=”Drugbank”,Class=”Pharmacological information”>
##SNV_DN=<Description = “Drug name(DN)”, Reference database=”Drugbank”,Class=”Pharmacological information”>
##SNV_DT=<Description = “Drug type(DT),Drugs are categorized by type, which determines their origin (Small molecule or Biotech)”, Reference database=”Drugbank”,Class=”Pharmacological information”>
##SNV_PGT=<Description = “Pharmacological gene type,It consists of four types: target, enzyme, transporter, carrier”, Reference database=”Drugbank”,Class=”Pharmacological information”>
##PROTEIN_Uniplot=<Description = “Uniplot identifier”, Reference database=”Uniplot”,Class=”Protein”>
##PROTEIN_Nextprott=<Description = “NextProt identifier”, Reference database=”NextProt”,Class=”Protein”>
##PROTEIN_PDB=<Description = “Protein Data Bank(PDB) identifier”, Reference database=”PDB”,Class=”Protein”>
##PROTEIN_Enemble_Pro=<Description = “Ensemble protein(Pro) identifier”, Reference database=”Ensemble”,Class=”Protein”>
##TRANSCRIPT_Enemble_Tra=<Description = “Ensemble transcript(Tra) identifier”, Reference database=”Ensemble”,Class=”Transcript”>
##GENE_Enemble_Gen=<Description = “Ensemble Gene(Gen) identifier”, Reference database=”Ensemble”,Class=”Gene”>
##GENE_GF=<Description = “Gene Families(GF)”, Reference database=”Ensemble”,Class=”Gene”>
##GENE_GD=<Description = “Gene description(GD)”, Reference database=”HGNC”,Class=”Gene”>
##GENE_GS=<Description = “Gene Symbol(GS)”, Reference database=”HGNC”,Class=”Gene”>
##GENE_HGNC=<Description = “HGNC ID”, Reference database=”HGNC”,Class=”Gene”>
##GENE_UCSC=<Description = “UCSC ID”, Reference database=”UCSC”,Class=”Gene”>
##GENE_Cosmic=<Description = “Cosmic Gene ID”, Reference database=”Cosmic”,Class=”Gene”>
##GENE_Entrez=<Description = “Entrez ID”, Reference database=”Ensemble”,Class=”Gene”>
##GENE_ RefSeq=<Description = “Reference Sequence (RefSeq) accession number”, Reference database=”Entrez”,Class=”Gene”>
##Disease_Omim=<Description = “Omim ID”, Reference database=”Omim”,Class=”Clinical information”>
##DRUG_PharmGKB=<Description = “PharmGKB ID”, Reference database=”PharmGKB”,Class=”Pharmacological information”>
##DRUG_CHEMBLI=<Description = “CHEMBL ID”, Reference database=”CHEMBLI”,Class=”Pharmacological information”>
##Literature_PMID=<Description = “Pubmed ID”, Reference database=”Pubmed”,Class=”Literature”>
##Biological function_STRING=<Description = “STRING ID”, Reference database=”STRING”,Class=”Biological function”>
##Biological function_Vega=<Description = “Vega ID”, Reference database=”Vega”,Class=”Biological function”>
##Biological function_ENA=<Description = “European Nucleotide Archive ID”, Reference database=”ENA”,Class=”Biological function”>
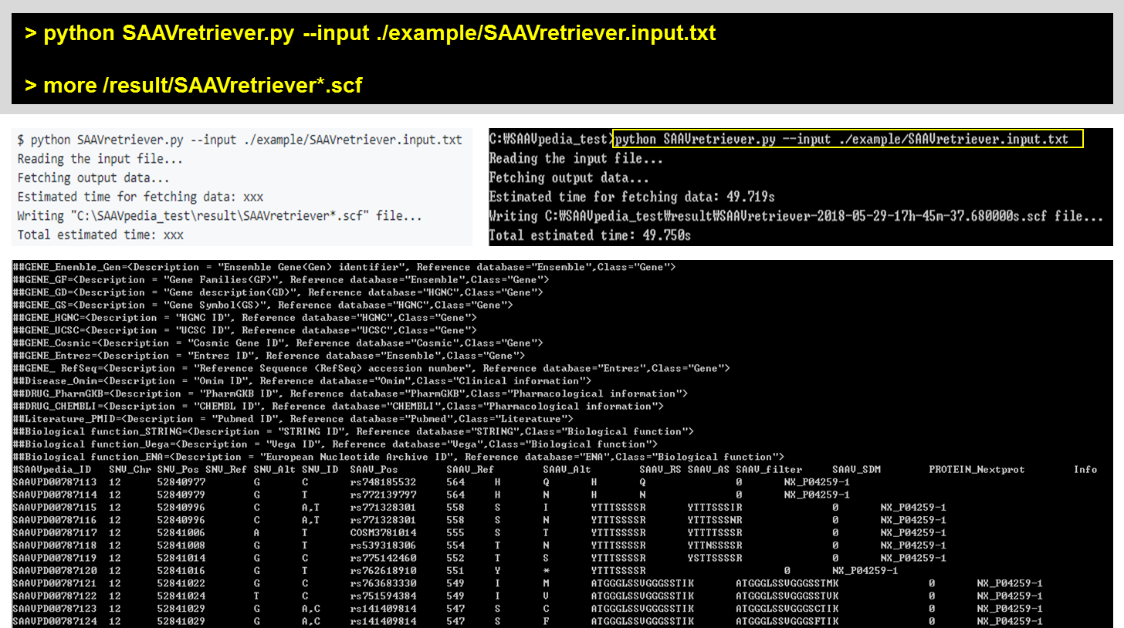
#SAAVpedia_ID SNV_Chr SNV_Pos SNV_Ref SNV_Alt SNV_ID SAAV_Pos SAAV_Ref SAAV_Alt SAAV_RS SAAV_AS SAAV_filter SAAV_SDM PROTEIN_Uniprot Info SAAV_QS SAAV_Pvalue SAAVPD02593079 1 151238552 G T COSM5823395 243 A S AAAASAAEAGIATTGTEDSDDALLK AAAASSAEAGIATTGTEDSDDALLK FAIL 1 P55036 1.2819 0.398801 SAAVPD00180028 10 76154005 C T rs762559805 92 A V AAEAHVDAHYYEQNEQPTGTCAACITGDNR AAEAHVDVHYYEQNEQPTGTCAACITGDNR PASS 1 P55263 2.2861 0.036260 SAAVPD01684812 17 1257591 C T rs762403227 188 S N AAFDDAIAELDTLSEESYK AAFDDAIAELDTLNEESYK FAIL 0 P62258 0.7440 0.714988 SAAVPD01960959 17 76970895 C T rs762942693 84 G D AAFGQGSGPIMLDEVQCTGTEASLADCK AAFGQGSDPIMLDEVQCTGTEASLADCK FAIL 0 Q08380 0.7058 0.734564 SAAVPD02833306 1 22168801 T G rs2229491 2995 S G AASGPGPEQEASFTVTVPPSEGSSYR AAGGPGPEQEASFTVTVPPSEGSSYR PASS 1 P98160 2.6799 0.008207
3.2.2.1 Meta-information
Meta-information is included after the ## string with key and value pairs. The value consists of the description, reference database, and functional class in SAAVpedia.
3.2.2.1.1 File format
A single “fileformat” field is always required in the first line, including the SCF format version number.
##fileformat=SCFv1.0
3.2.2.1.2 File date
“filedate” indicates the date the file was created.
##fileDate=20180529
3.2.2.1.3 SAAVpedia sequence database
“SAAVpedia sequence database” includes the reference database version of neXtProt.
##SAAVpedia sequence database=neXtprot Jan 2017
3.2.2.1.3 Functional information
The information lines describing the functional entries used in the SCF-AF file are included in the meta-information section.
##Functional information
##SNV_OID=Description = <“EFO ontology ID(OID) of SNV_Phenotype.”, Reference database=”EFO”,Class=”Clinical information”>
3.2.2.2 Header line syntax
The header line has 15 fixed columns. These columns are as follows:
1. SAAVpedia ID
2. SNV_Chr
3. SNV_Pos
4. SNV_Ref
5. SNV_Alt
6. SNV_ID
7. SAAV_Pos
8. SAAV_Ref
9. SAAV_Alt
10. SAAV_RS
11. SAAV_AS
12. SAAV_Filter
13. PROTEIN_neXtProt
14. SAAV_SDM
15. INFO
3.2.2.3 Data lines
There are 15 fixed fields per record. All data lines are tab-delimited.
Table 1. Symbols in SCF and their descriptions.
|
SAAVpedia ID |
Internal identifier of SAAV. |
|
SNV_Chr |
Single nucleotide variant chromosome |
|
SNV_Pos |
Single nucleotide variant position |
|
SNV_Ref |
Single nucleotide variant reference base |
|
SNV_Alt |
Single amino acid variant alteration base |
|
SNV_ID |
Genomic variation reference ID (dbSNP, COSMIC) |
|
SAAV_Pos |
Single amino acid variant position |
|
SAAV_Ref |
Single amino acid variant reference base |
|
SAAV_Alt |
Single amino acid variant alteration base |
|
SAAV_RS |
Peptide reference sequence( RS) that contains |
|
SAAV_AS |
Peptide alteration sequence (AS) that |
|
SAAV_Filter |
filter status, PASS if this SAAV has less than 0.05 SAAV_Pvalue. If not, then FAIL |
|
PROTEIN_neXtProt |
neXtProt identifier |
|
SAAV_SDM |
Strand direction matching information of SNV and SAAV. If the information is correct, the score is “1”, otherwise “0” |
|
INFO |
Information |
4. Usage
4.1 Test data
The test data include two kinds of datasets, from breast cancer and glioblastoma. The SAAV test data for SAAVidentifier, SAAVretriever, and SAAVannotator was obtained from the CPTAC breast cancer study. The SNV test data for SNVretriever was obtained from TCGA and ICGC. We extracted small subsets from the breast cancer and glioblastoma datasets for a demonstration of SAAVpedia.
4.1.1 Input file for SAAVidentifier
The input file for the SAAVidentifier module consists of 1,000 of 15,225 SAAV peptides extracted from the breast cancer dataset.

4.1.2 Input files for SAAVannotator and SAAVvisualizer
SAAVannotator and SAAVvisualizer accept SCF files as input files from SAAVidentifier. In this example, we provided two input files for SAAVannotator and SAAVvisualizer.
SAAVannotator.input.scf and SAAVvisualizer.input.scf files consist of 15 columns, as shown in Table 1.
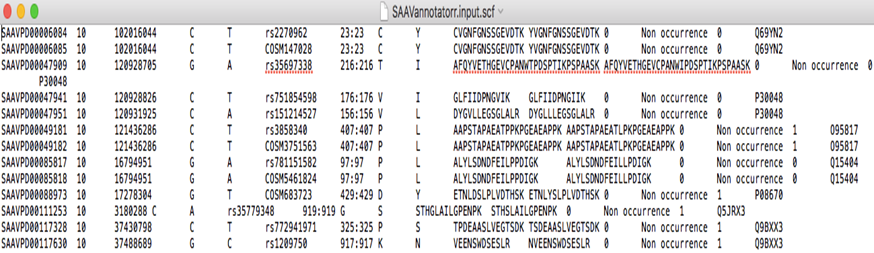
4.1.3 Input file for SNVretriever
The SNVretriever accepts text files consisting of dbSNP or COSMIC ID as input files.
SNVretriever.input.txt file is a list of dbSNP or COSMIC ID.

4.1.4 Input file for SAAVretriever
The SAAVretriever accepts text files consisting of Ensembl identifiers (ENSG, ENST, and ENSP IDs), UnitProt and neXtProt accession numbers, human chromosome numbers, and single amino acid variant positions, as input files, as as shown below. “chr:###” and “AApos:###” are defined as the inputs of chromosome number and amino acid position, respectively. The “###” indicates a positive integer value.
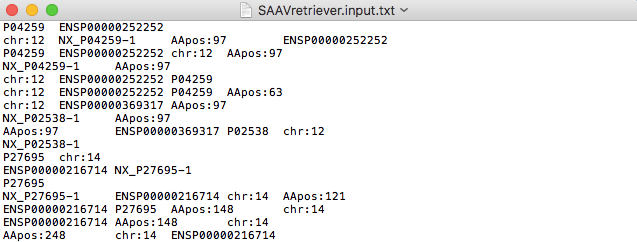
4.2 Proteogenomic interpretation pipeline
SAAVpedia provides a proteogenomic analysis pipeline to reduce and interpret SAAVs. The pipeline contains a database for proteome searches, a SAAV quality checker, a query builder based on clinical, biological, and pharmacological information, and a retrieving and interpreting system. SAAVpedia also provides a nsSNV and SAAV retriever pipeline to find SAAVs in the same location as input nsSNVs or identified from different proteomic data of various phenotype conditions.
The SAAVpedia is a comprehensive proteogenomic interpretation platform that identifies, annotates, and retrieves SAAVs from proteomic and genomic sequence data to discover true pathogenic variant candidates. SAAVpedia provides a proteogenomic analysis pipeline to reduce and interpret SAAVs. The pipeline contains a database for proteome searches, a SAAV quality checker, a query builder based on clinical, biological, and pharmacological information, and a retrieving and interpreting system.
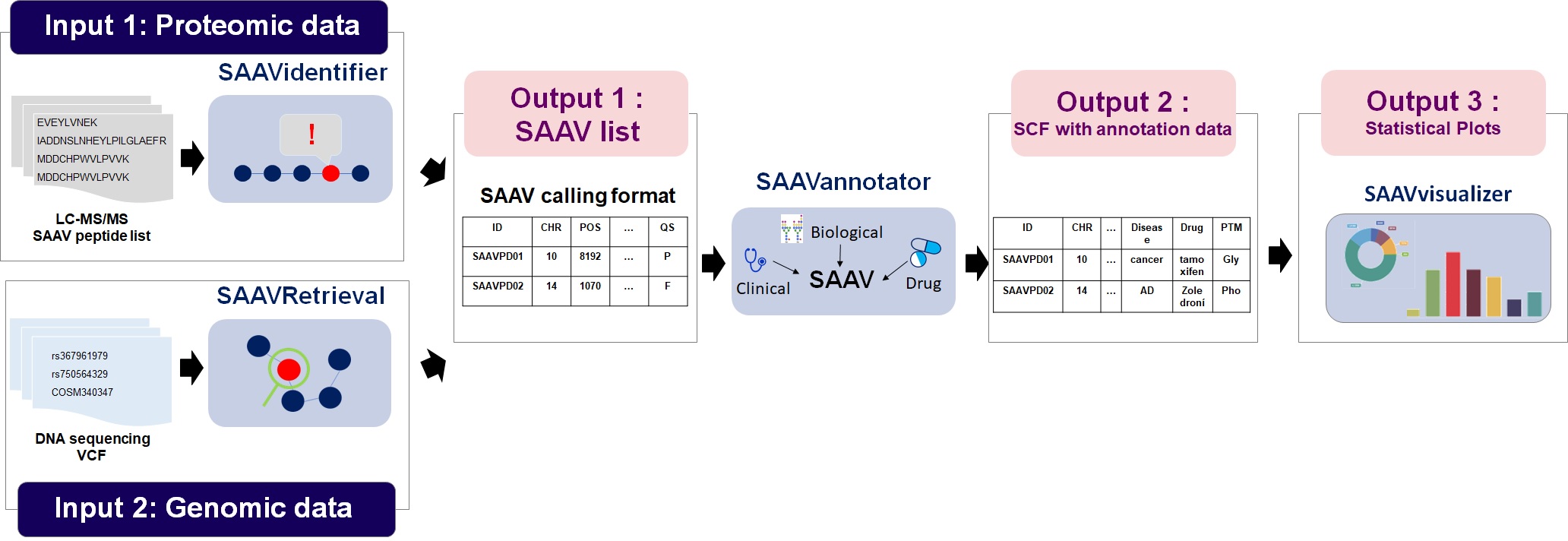
*SCF: SAAV-calling format; SCF-FI: SCF with functional information
4.3.1 SAAVidentifier module
I. Click “SAAVidentifier” in the main menu of the SAAVpedia hompage.
II. Check the latest database version.
III. Click “Download the example file”.
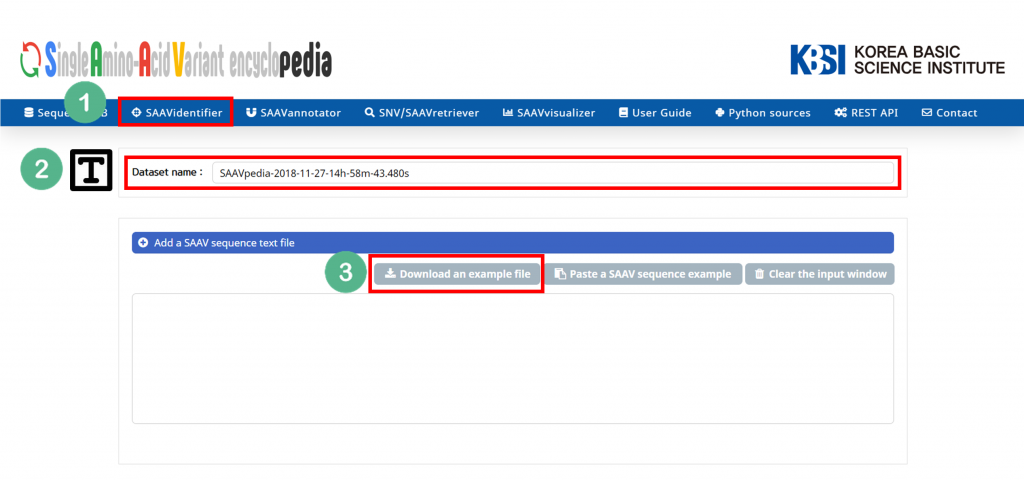
IV. Open the example file and copy the SAAV peptide sequences.
Then, paste the SAAV peptide sequences in the window.
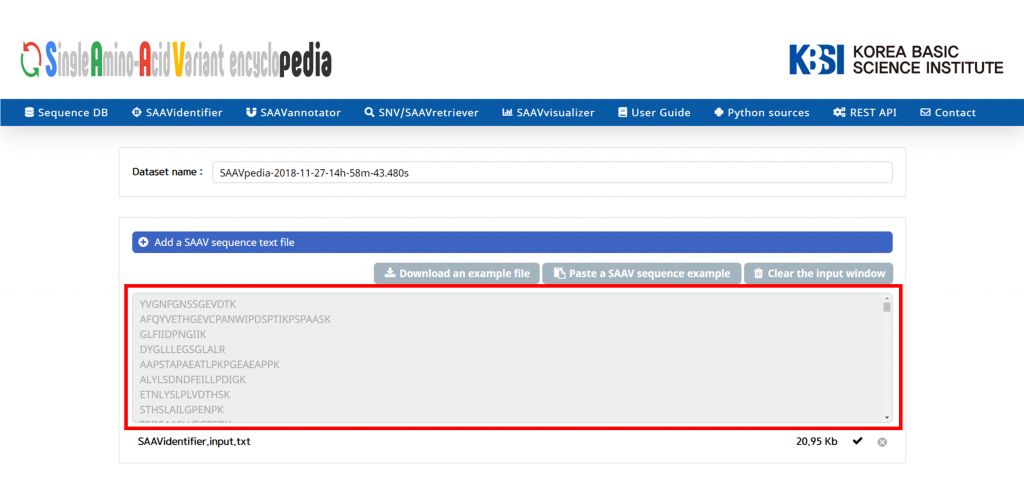
V. Click and download the example DTASelect-filter.txt file.
VI. Click the banner ‘Add “DTASelect-filter.txt” file(s)’.
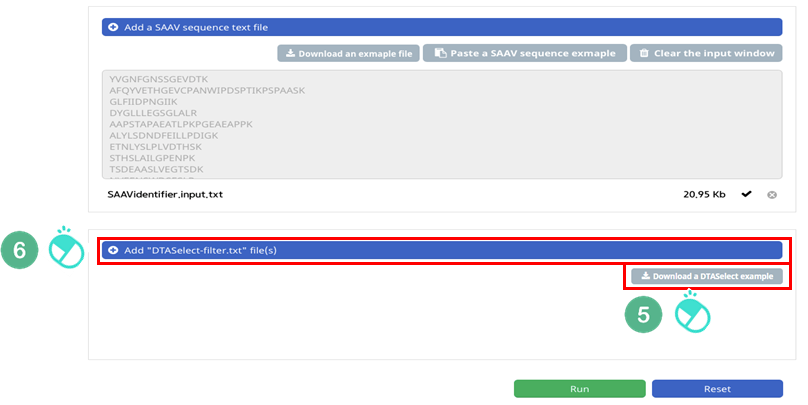
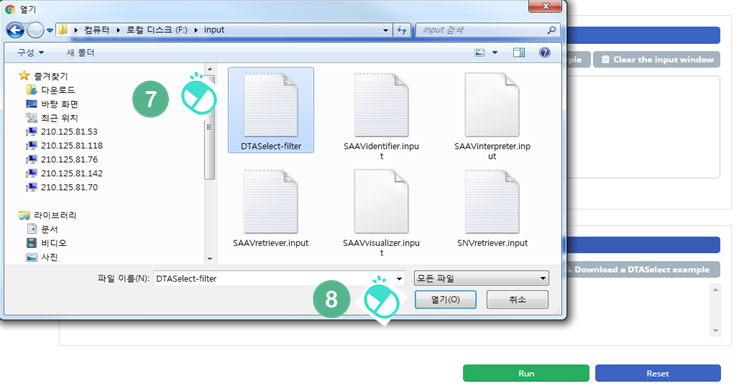
VII. Select the downloaded DTASelect-filter.txt file.
VIII. Click the “Open” button.
IX. Check the DTASelect-filter.txt file and click “Run”.
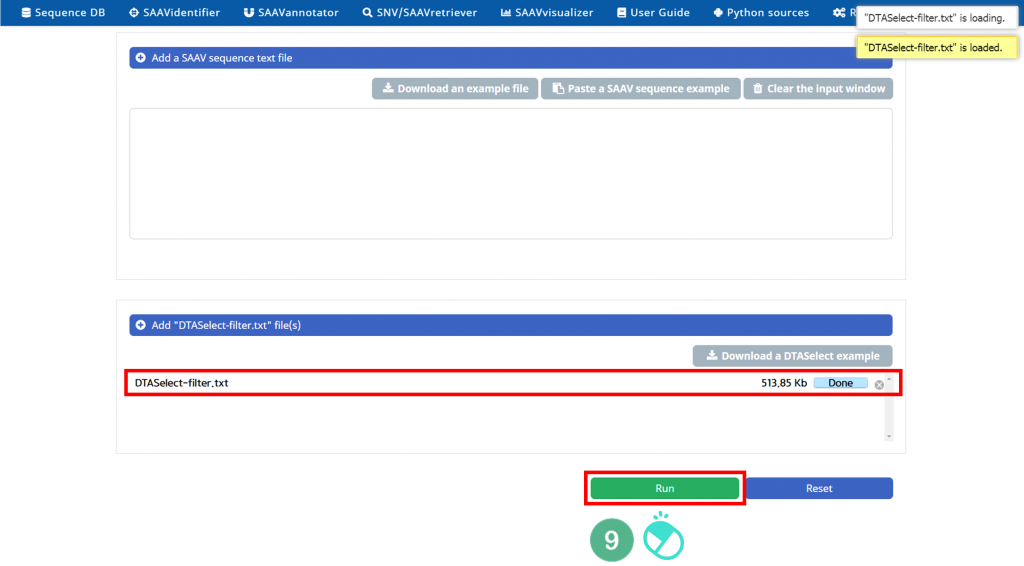
X. After running the SAAVidentifier module, the SCF data table of identified SAAVs will be shown in the web browser. The results page has two export buttons to save the file as an SCF or Excel file. The red and blue boxes present buttons to export the files in SCF or Excel format, respectively.
XI. SAAVvisualizer provides six kinds of statistical plots of identified SAAVs. Click each plot, and the plot will pop up.
XII. Click the “Next to SAAVannotator” button.
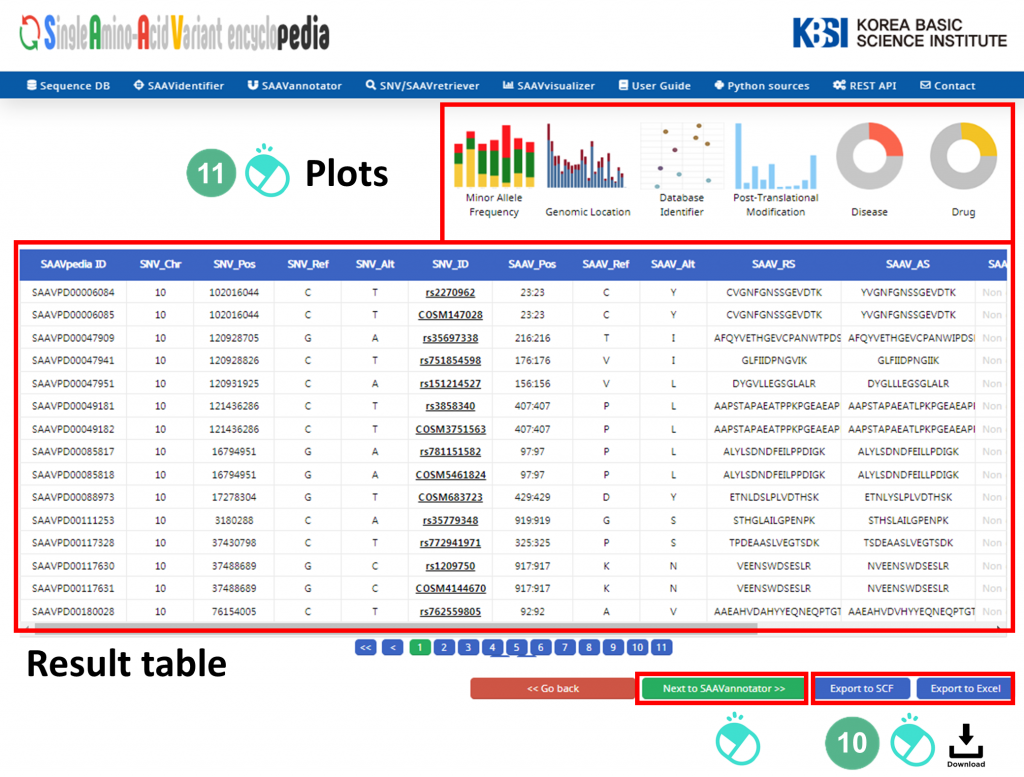
4.3.2 SAAVannotator module
I. Click “SAAVannotator” in the main menu.
II. Click and download an example SCF file.
III. Click “Add a SCF file”.

IV. Select the downloaded SCF file.
V. Click “Open”.

The SCF file is added to the input window.
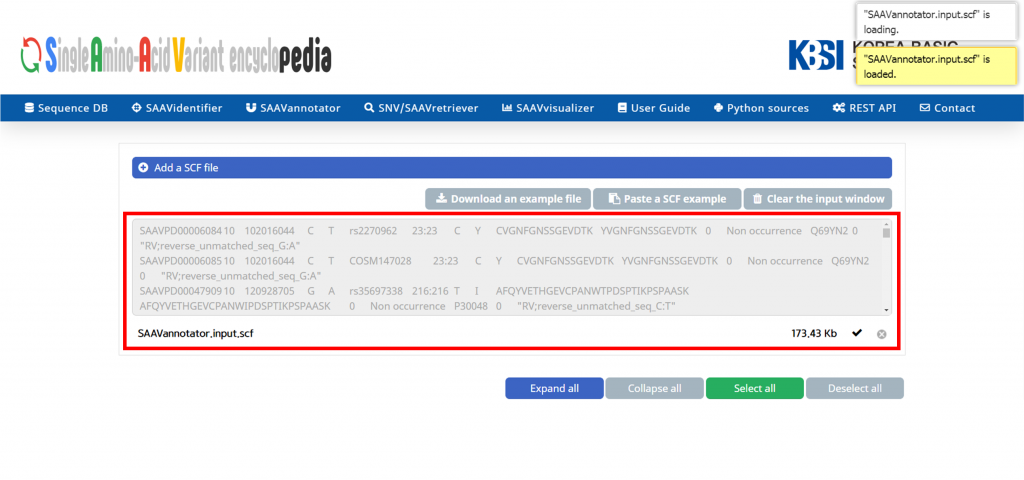
V. Click and download the example DTASelect-filter.txt file.
VI. Click the banner ‘Add “DTASelect-filter.txt” file(s)’.
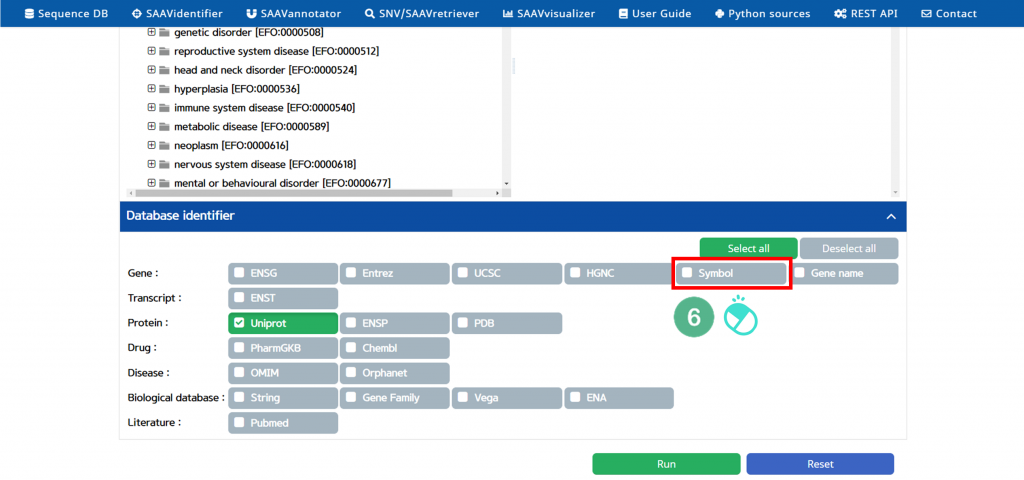
VI. Click the “Symbol” button.
The user can choose any other database category.
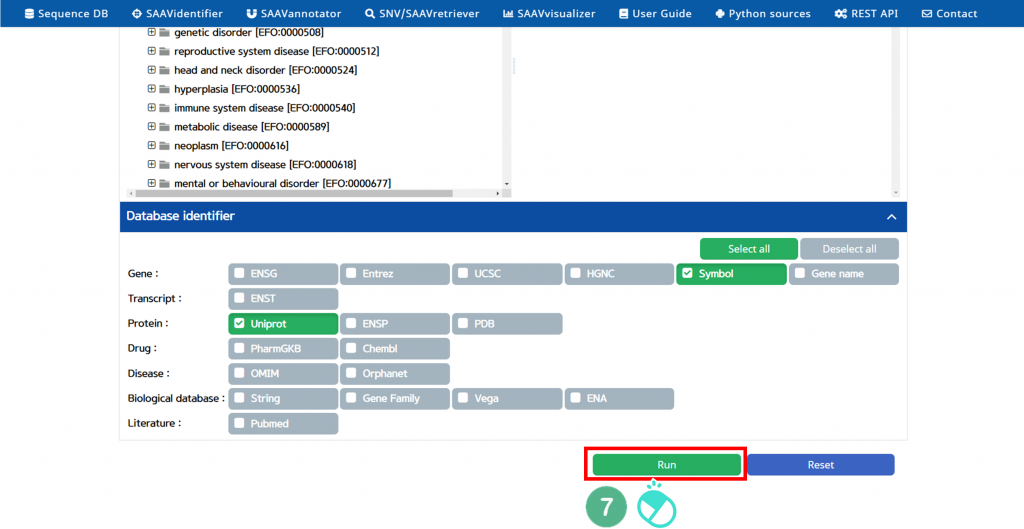
VII. Click “Run”.
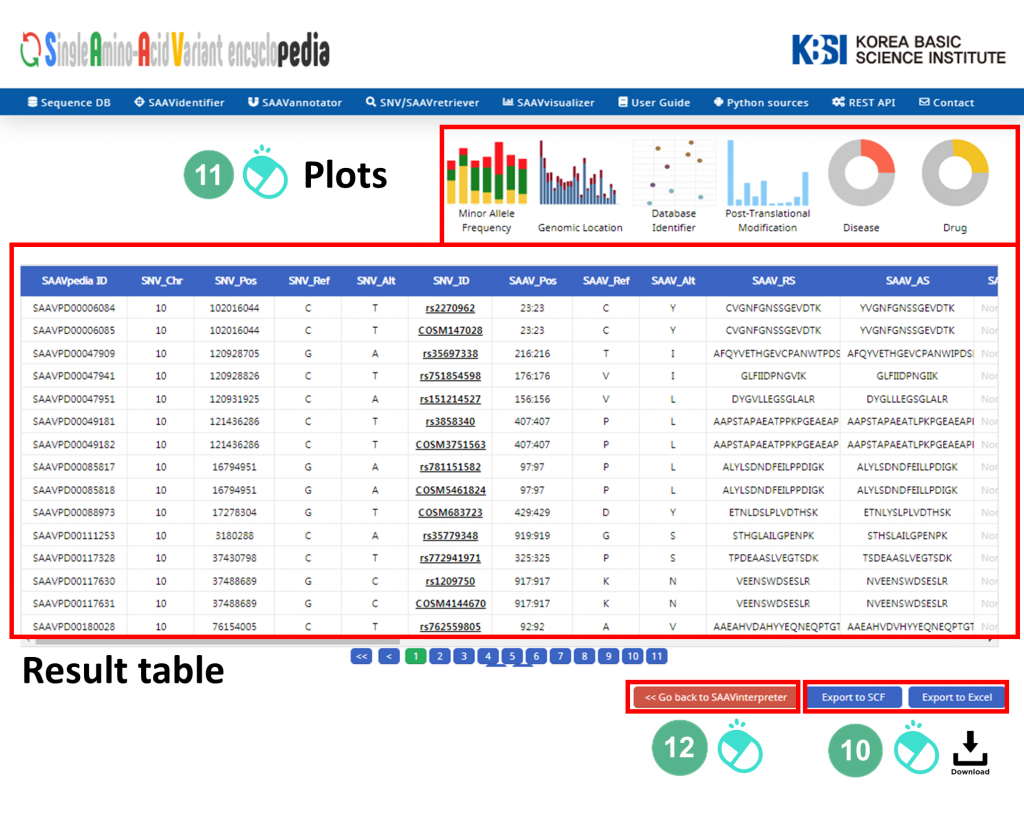
VIII. After running the SAAVannotator module, the SCF data table of annotated SAAVs will be shown in the web browser. The results page has two export buttons to save the resulting SCF or Excel file. The red and blue boxes present buttons to export files in SCF or Excel format, respectively.
IX. SAAVvisualizer provides six kinds of statistical plots of identified SAAVs. Click each plot, and the plot will pop up.
4.3.3 SNVretriever module
I. Click “SNV/SAAVretriever” in the main menu.
II. Check the latest database version.
III. Click “SNVretriever”.
IV. Download the example file.
V. Click “Add a SNV/SAAVretriver input file”.
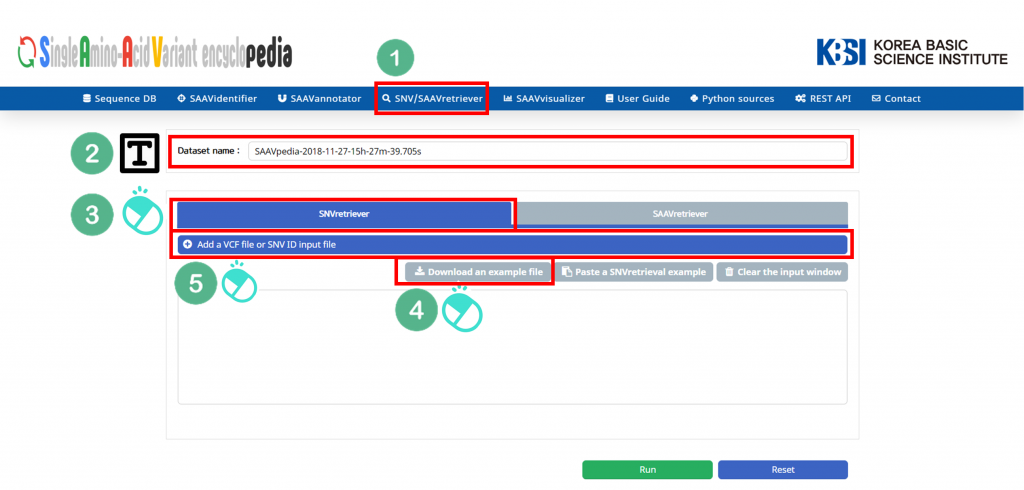
VI. Select the downloaded file.
VII. Click “Open”.

The dbSNP or COSMIC identifier is shown in the input window.
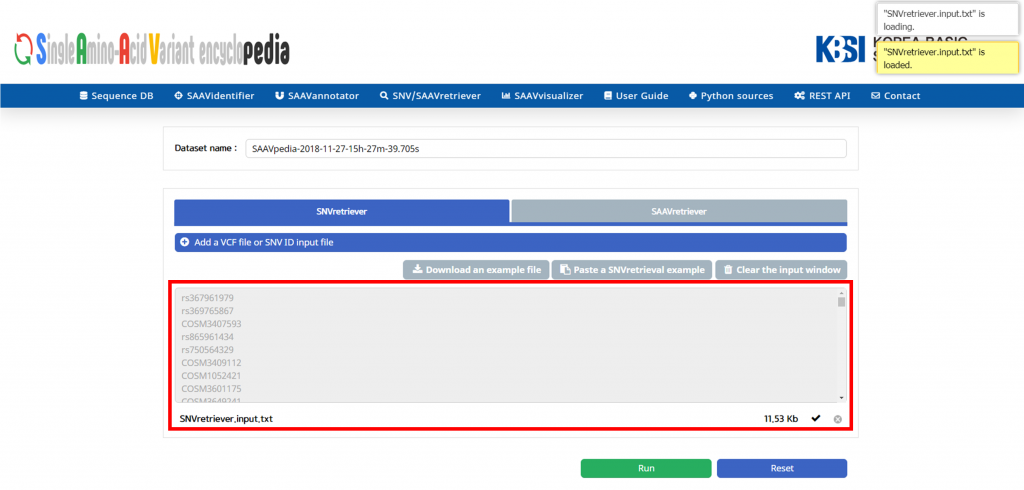
VIII. Click “Run”.
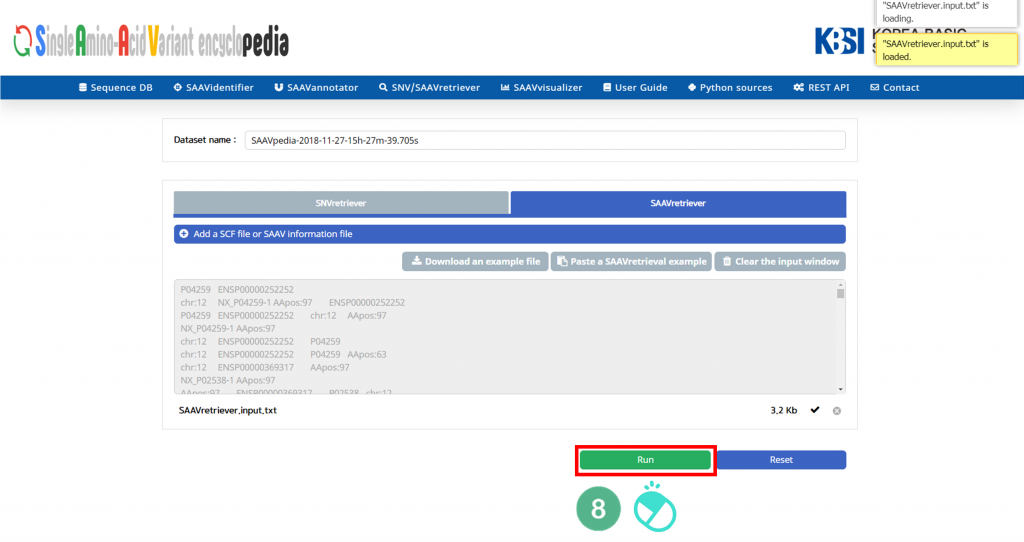
IX. After running the SNVretriever module, the SCF data table of annotated SAAVs will be shown in the web browser. The results page has two export buttons to save the resulting SCF or Excel file. The red and blue boxes present buttons for exporting files in SCF and Excel format, respectively.
X. SAAVvisualizer provides six kinds of statistical plots of identified SAAVs. Click each plot, and the plot will pop up.
Click “SAAVannotator”.

4.3.4 SAAVretriever module
I. Click “SNV/SAAVretriever” in the main menu.
II. Check the latest database version.
III. Click “SAAVretriever”.
IV. Download the example file.
V. Click “Add a SNV/SAAVretriever input file”.
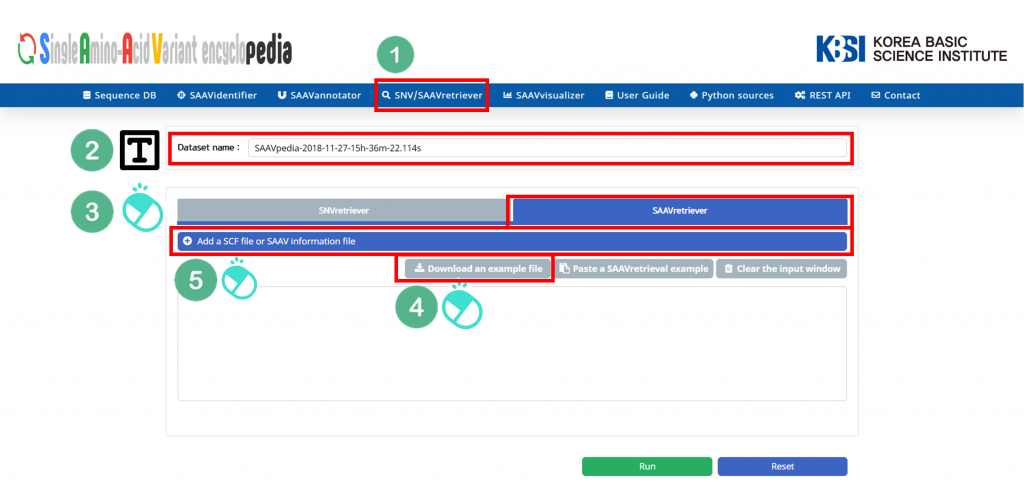
VI. Select the downloaded file.
VII. Click “Open”.
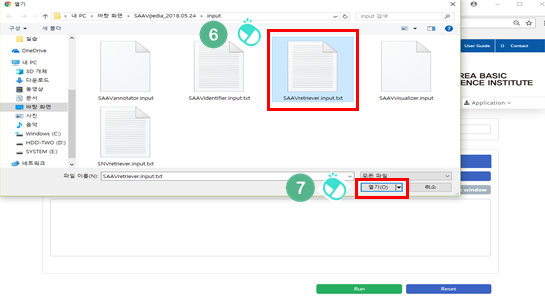
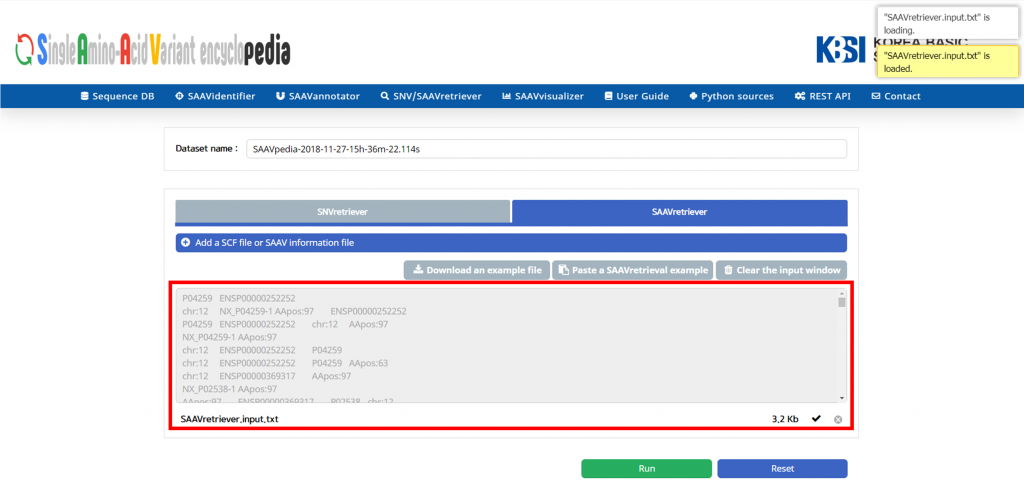
VIII. Click “Run”.
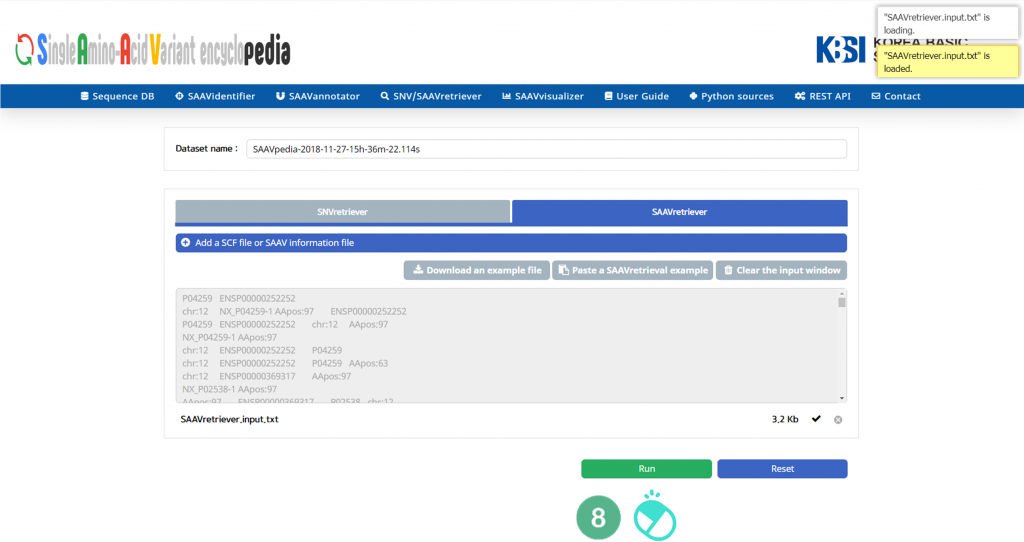
IX. After running the SAAVannotator module, the SCF data table of annotated SAAVs will be shown in the web browser. The results page has two export buttons to save the resulting SCF or Excel file. The red and blue boxes present buttons to export files in SCF and Excel format, respectively.
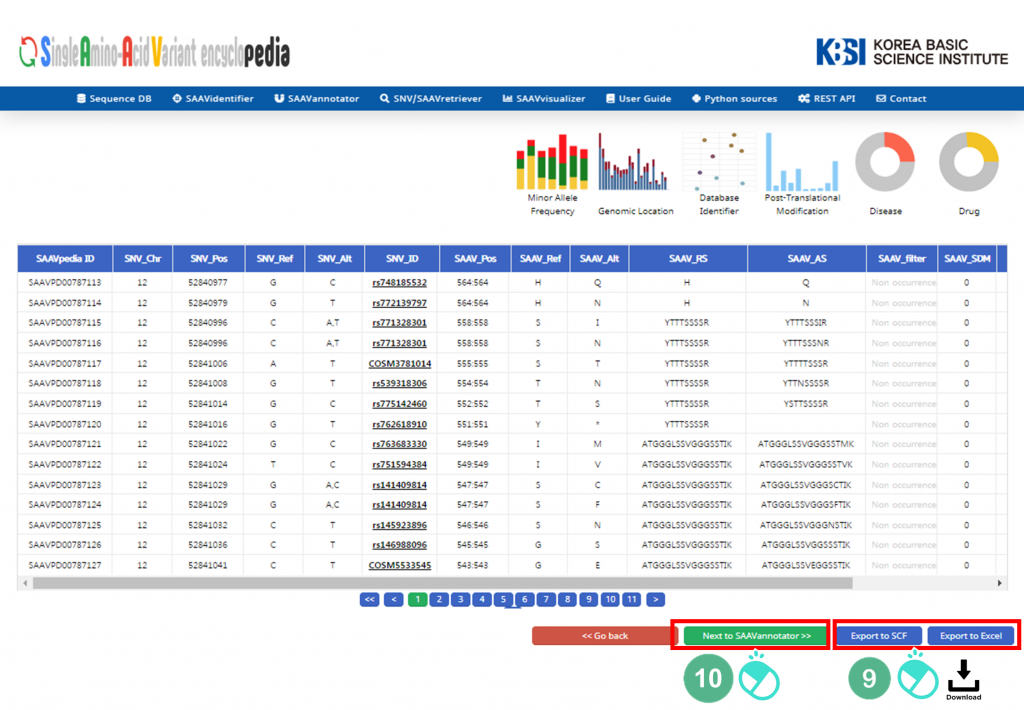
X. SAAVvisualizer provides six kinds of statistical plots of identified SAAVs. Click each plot, and the plot will pop up.
XI. Click “SAAVannotator”.
4.3.5 SAAVvisualizer module
I. Click “SAAVvisualizer” in the main menu.
II. Click “Add a SCF file”.
III. Download the example file.
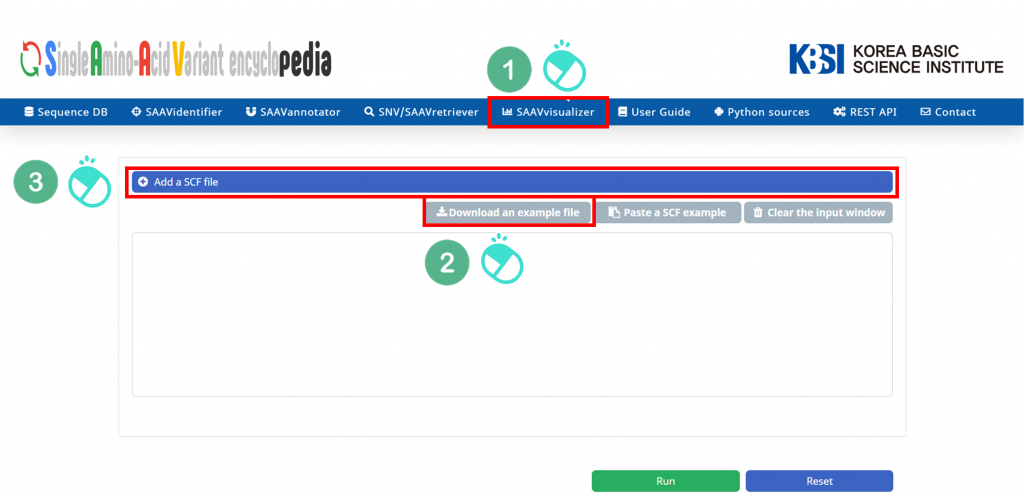
IV. Select the downloaded file, and click “Open”.
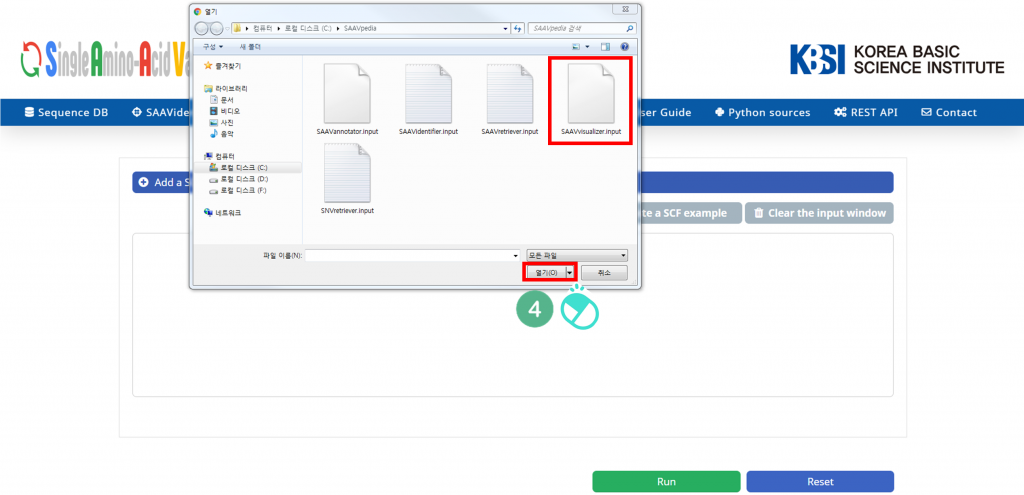
V. Click “Run”.

VI. After running the SAAVvisualizer module, six plot icons will be shown in the web browser.
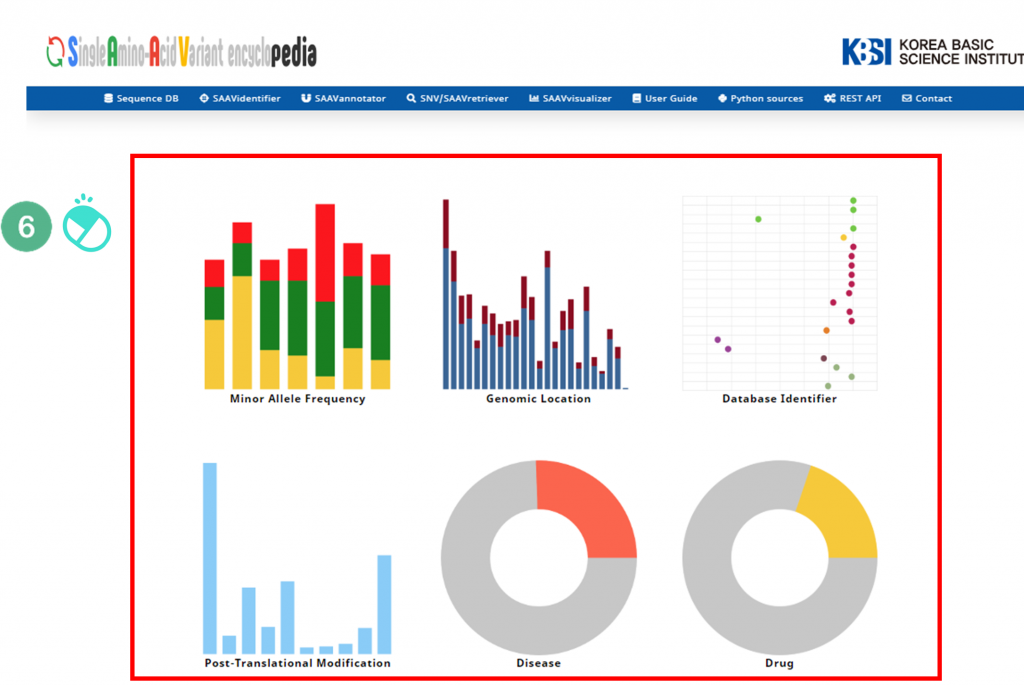
VII. Six kinds of statistical plots are provided.

4.4 Python stand-alone program
SAAVpedia offers a stand-alone program as a Python package for users who want to control large amounts of input data. The SAAVpedia Python package does not require an Internet connection after the Python package installation is complete. Recent SAAVpedia Python source code is freely available via the GitHub repository (https://github.com/saavpedia/python/). For details, please visit the GitHub repository.
4.4.1 Installation of the SAAVpedia Python package
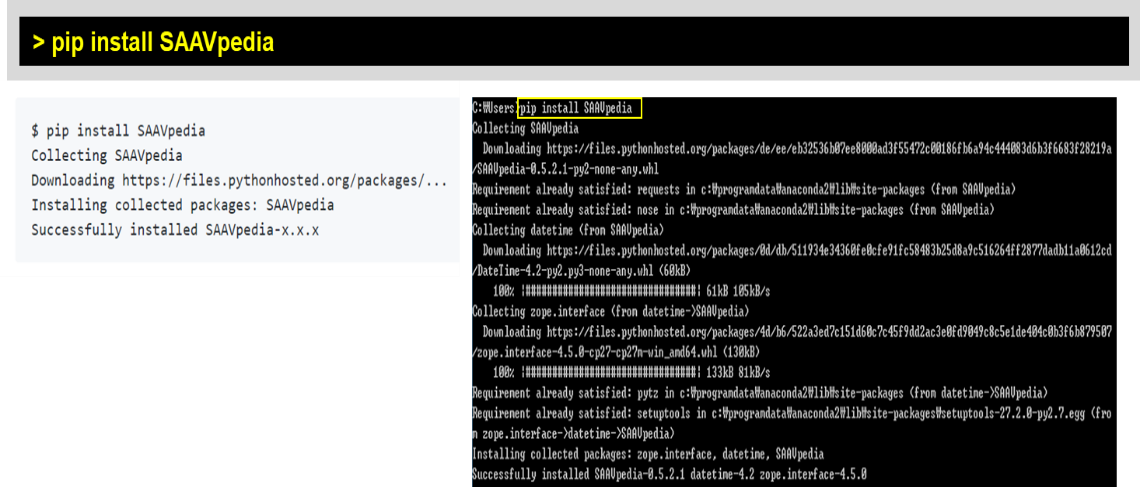
Python is freely available as an open source tool on all major operating systems including MS Windows, MAC OS, and Linux. Recent Python versions (starting with Python 2.7.9 and 3.4) include the Python package management tool (pip), which allows easy installation from the command line. SAAVpedia offers a standalone program via the Python Package Index, which is a repository of libraries for the Python programming language (https://pypi.org/project/saavpedia/). Therefore, the SAAVpedia Python package can be installed as shown below:
> pip install SAAVpedia
4.4.1.1 Download the SAAVpedia database
To use SAAVpedia without the Internet, you must download a SAAVpedia database file. After installing the SAAVpedia Python package, you can download the SAAVpedia database file to the Python library path as shown below:
> python
>>> from SAAVpedia import SAAVpedia
>>> SAAVpedia().init()
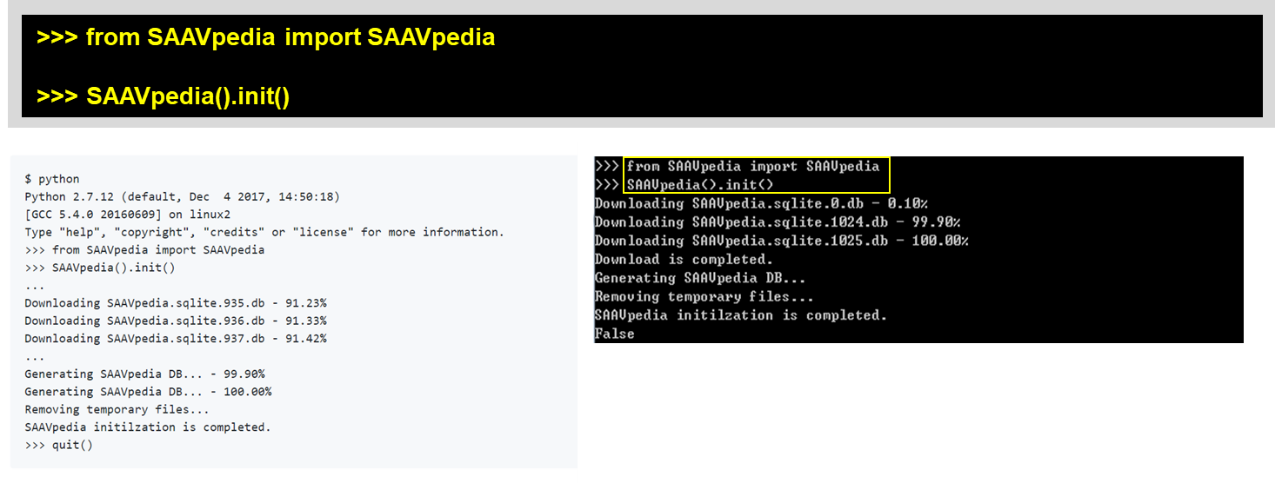
If the download speed is too slow in the initialization process, visit the SAAVpedia download website (https://www.saavpedia.org/python-sources/). We provide Python source codes with the SAAVpedia database file. Download the “Full version”.
4.4.1.2 Installation of Proteogenomic Interpretation Pipelinee
To support the proteogenomic interpretation pipeline, the SAAVpedia Python package provides four command scripts, “SAAVidentiifer.py”, “SAAVannotator.py”, “SAAVretriever.py”, and “SNVretriever.py”, with example files for demonstration. To obtain these files, import the SAAVpedia module in Python and run its install function. This install function needs a parameter on the destination path to copy command scripts. The details of installing SAAVpedia command scripts are shown below.
> python
>>> from SAAVpedia import SAAVpedia
>>> SAAVpedia().install(‘Destination path’)

4.4.2 Use of the Proteogenomic Interpretation Pipeline
After installation is complete, SAAVpedia module scripts and their example files are located in the destination folder.
4.4.2.1 SAAVidentifier.py
The “SAAVidentifier.py” file is a Python script file for identifying SAAVs from SAAV peptides. In the “example” folder, the name of the input file is “SAAVidentifier.input.txt”. The “–input” option is used to present the path of a SAAV peptide list file. The “–output” option represents an SCF file path for saving the output. When the output option is ignored, a result file will be automatically generated in the “result” folder.
To print help messages, type “python SAAVidentifier.py” as below:
> python SAAVidentifier.py

For an example:
> python SAAVidenfier.py –input ./example/SAAVidentifier.input.txt
4.4.2.2 SAAVannotator.py
The input “SAAVannotator.py” accepts a result SCF file generated by the SAAVidentifier and SNV/SAAVretriever modules. This module has more than 40 options for annotating functional information including 1,000G MAF, ESP MAF, variant type, phenotype, Ensembl identifiers, EFO ID, various databases, etc. For more details, type “python SAAVannotator.py” in the command line. The example file is “SAAVannotator.input.scf”.
To print SAAVannotator options, type “python SAAVannotator.py” as below:
> python SAAVannotator.py
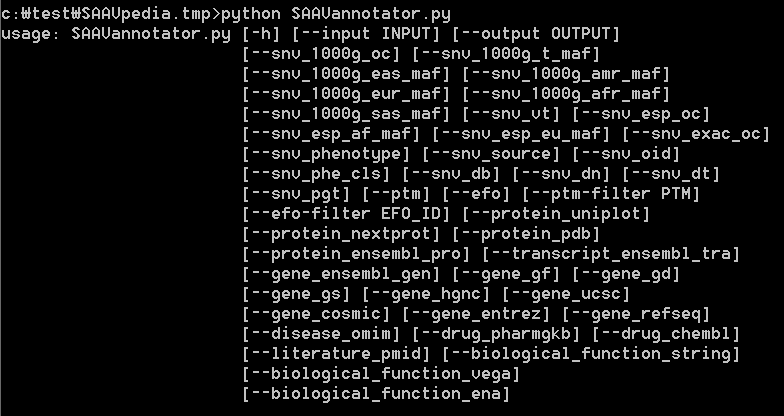
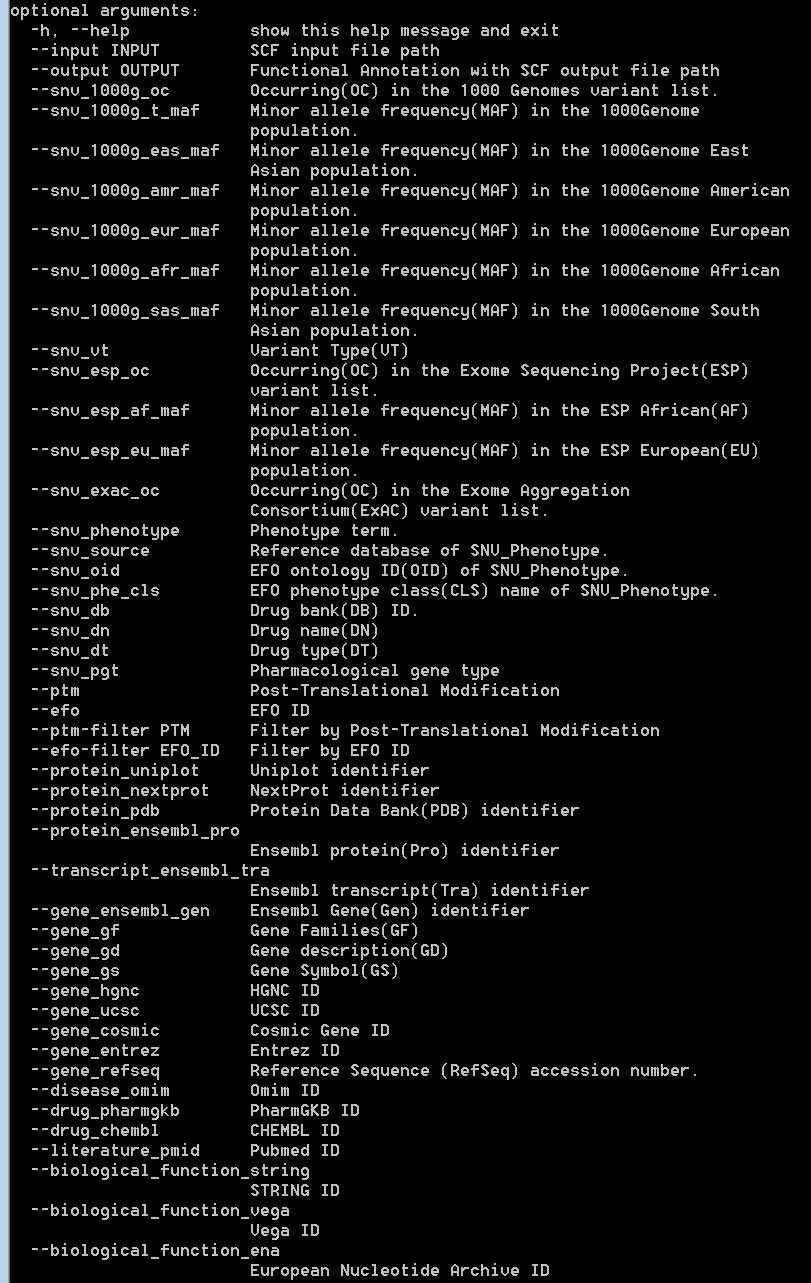
|
SAAVannotator option |
Description |
|
–snv_1000g_oc |
Occurring (OC) in the 1000Genome variant list. |
|
–snv_1000g_t_maf |
Minor allele frequency (MAF) in the 1000Genome population. |
|
–snv_1000g_eas_maf |
Minor allele frequency (MAF) in the 1000Genome East Asian population. |
|
–snv_1000g_amr_maf |
Minor allele frequency (MAF) in the 1000Genome American population. |
|
–snv_1000g_eur_maf |
Minor allele frequency (MAF) in the 1000Genome European population. |
|
–snv_1000g_afr_maf |
Minor allele frequency (MAF) in the 1000Genome African population. |
|
–snv_1000g_sas_maf |
Minor allele frequency (MAF) in the 1000Genome South Asian population. |
|
—snv_vt |
Variant Type (VT) |
|
—snv_esp_oc |
Occurring (OC) in the Exome Sequencing Project (ESP) variant list. |
|
—snv_esp_af_maf |
Minor allele frequency (MAF) in the ESP African (AF) population. |
|
—snv_esp_eu_maf |
Minor allele frequency (MAF) in the ESP European (EU) population. |
|
—snv_exac_oc |
Occurring (OC) in the Exome Aggregation Consortium (ExAC) variant list. |
|
—snv_phenotype |
Phenotype term. |
|
—snv_source |
Reference database SNV_Phenotype. |
|
—snv_oid |
EFO ontology ID (OID) of SNV_Phenotype. |
|
—snv_phe_cls |
EFO phenotype class (CLS) name of SNV_Phenotype. |
|
—snv_db |
Drug bank (DB) ID. |
|
—snv_dn |
Drug name (DN) |
|
—snv_dt |
Drug type (DT) |
|
—snv_pgt |
Pharmacological gene type |
|
—ptm |
Post-Translational Modification |
|
—efo |
EFO ID |
|
—protein_uniplot |
Uniplot identifier |
|
—protein_nextprot |
NextProt identifier |
|
—protein_pdb |
Protein Data Bank (PDB) identifier |
|
—protein_ensembl_pro |
Ensembl protein (Pro) identifier |
|
—transcript_ensembl_tra |
Ensembl transcript (Tra) identifier |
|
—gene_ensembl_gen |
Ensembl Gene (Gen) identifier |
|
—gene_gf |
Gene Families GF) |
|
—gene_gd |
Gene Description (GD) |
|
—gene_gs |
Gene Symbol (GS) |
|
—gene_hgnc |
HGNC ID |
|
—gene_ucsc |
UCSC ID |
|
—gene_cosmic |
Cosmic Gene ID |
|
—gene_entrez |
Entrez ID |
|
—gene_refseq |
Reference Sequence (RefSeq) accession number |
|
—disease_omim |
Omim ID |
|
—drug_pharmgkb |
PharmGKB ID |
|
—drug_chembl |
CHEMBL ID |
|
—literature_pmid |
Pubmed ID |
|
—biological_function_string |
STRING ID |
|
—biological_function_vega |
Vega ID |
|
—biological_function_ena |
European Nucleotide Archive ID |
For an example:
If you want to annotate functional information, push the back option(s) to the SAAVannotator command. When “–snv_phenotype” and “–gene_gs” (Gene Symbol) options are used as a usage example of SAAVannotator, the command is as follows:
> python SAAVannotatr.py –input ./example/SAAVannotator.input.txt –snv_phenotype –gene_gs
4.4.2.3 SNV/SAAVretriever
There are SNVretriever.py and SAAVretriever.py files for searching SAAVs using nsSNV and SAAV information, respectively. The input of the SNVretriever module is the “SNVretriever.input.txt” file consisting of dbSNP and COSMIC IDs in the example folder.
[SNVretriever.py]
To print help messages for SNVretriever.py, type “python SNVretriever.py” as shown below:
> python SNVretriever.py
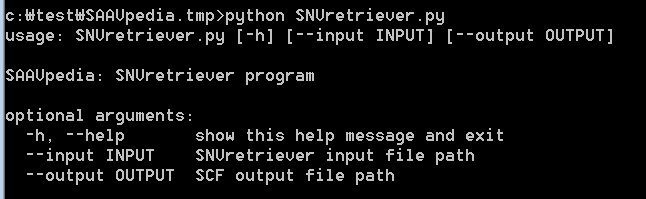
For an example:
> python SNVretriever.py –input ./example/SNVretriever.input.txt
[SAAVretriever.py]
To print help messages for SAAVretriever.py, type “python SAAVretriever.py” as shown below:
> python SAAVretriever.py

For an example:
> python SAAVretriever.py –input ./example/SAAVretriever.input.txt
4.5 REST API
SAAVpedia also provides REST API, which receives information on SAAVs with their functional annotation via standard HTML. The API returns outputs as XML and JSON formats. The WUI provides the REST Query Builder via a web page, https://www.saavpedia.org/rest-api/. The REST Query Builder consists of options for module, output, and example types, and two windows for inputs and outputs.
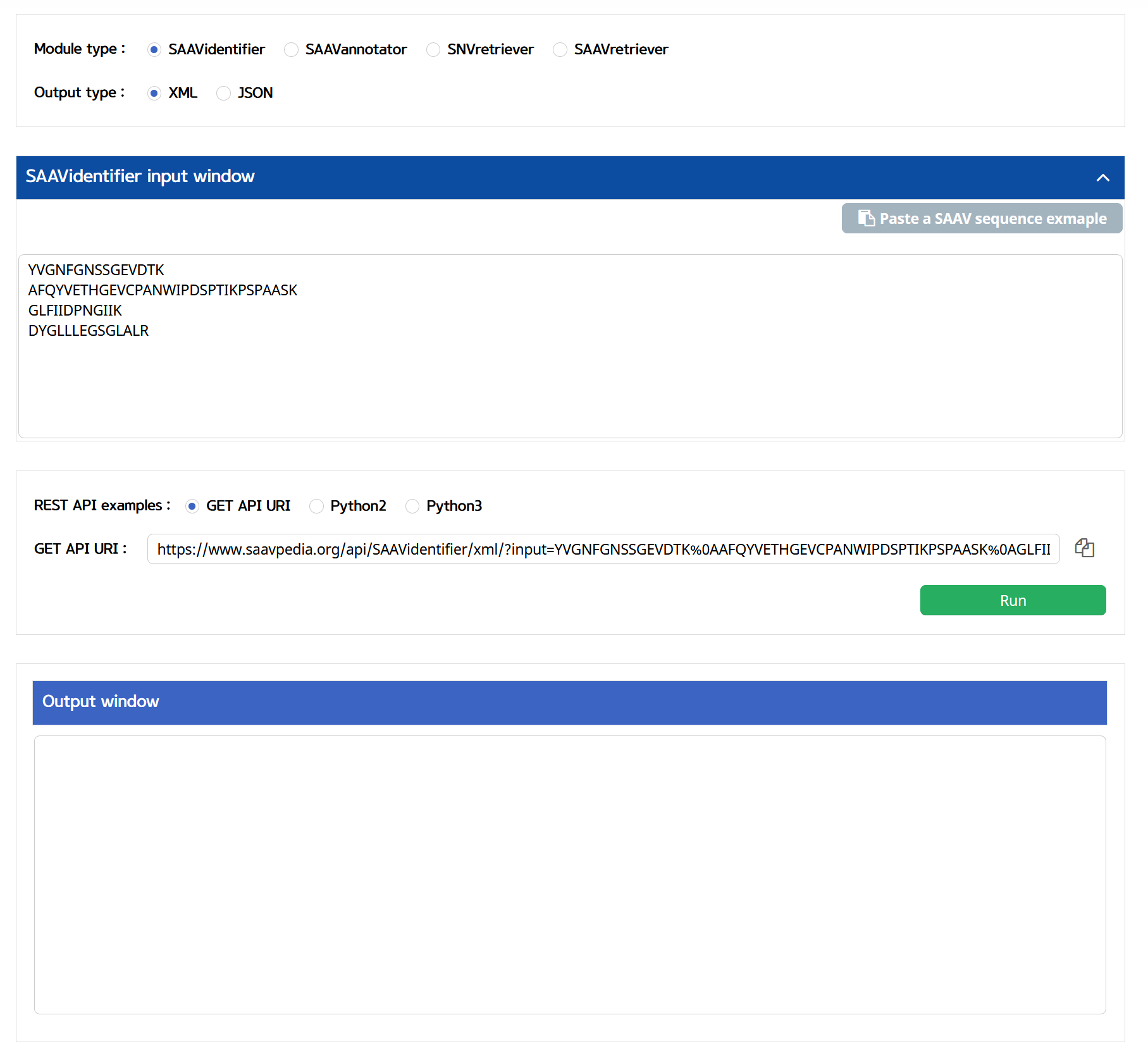
There are four module types, SAAVidentifier, SAAVannotator, SAAVretriever, and SNVretriever. The available output types are XML and JSON formats.
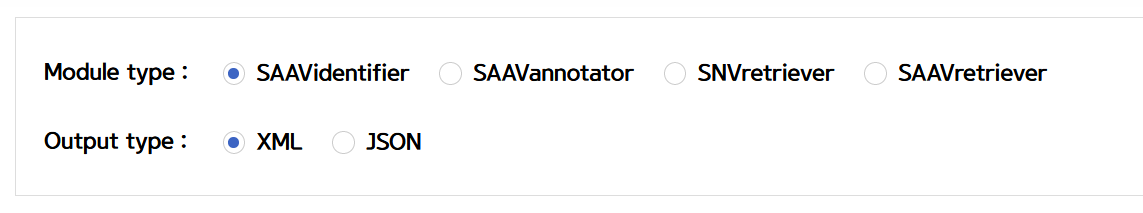
Builder also provides three REST API example types, GET API URI, Python2, and Python3 codes. Therefore, Python users can easily obtain sample code using the POST method.
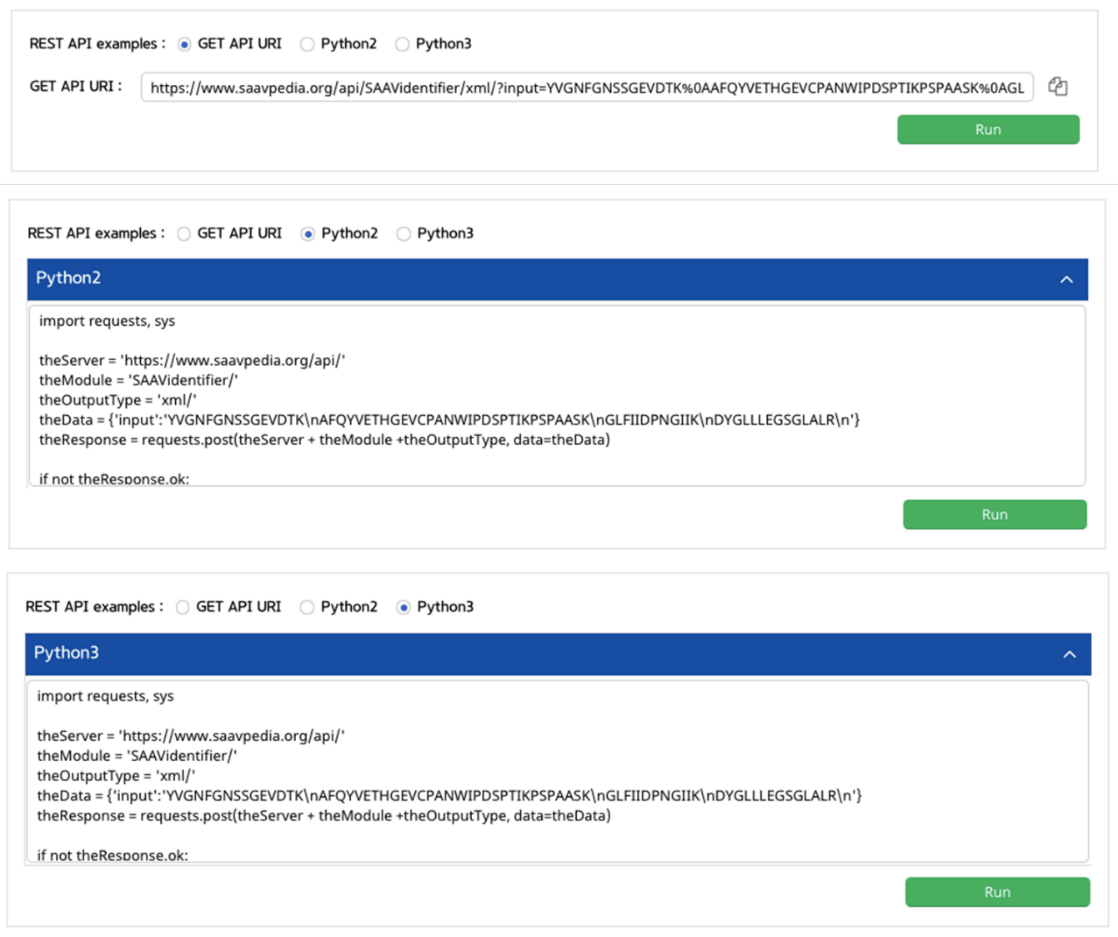
After running the REST Query Builder, you can easily see XML or JSON format output.
[XML output]

[JSON output]
5. User manual
5.1 Web interface panel
5.1.1 SAAV sequence database download
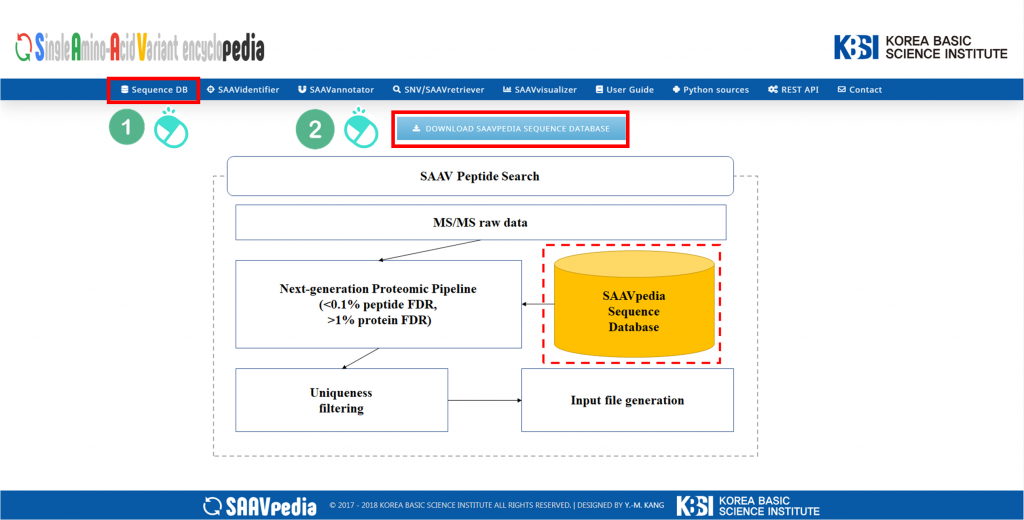
I. Click “Sequence DB”.
II. Click and download the SAAVpedia sequence database.
5.1.2 SAAVidentifier
The SAAVidentifier module provides a SAAV-calling format (SCF) file format, which includes the SAAV list, genomic and proteomic variant information using the SAAV reference database, and a quality score calculated from the proteome search results of each corresponding SAAV peptide. The SAAVidentifier web interface panel is as shown below:
I. Database Version.
II. File manager for opening and adding text files of SAAV peptide lists.
III. Button to download an example SAAV peptide list file.
IV. Button to automatically paste example SAAV peptide sequences.
V. Button to clear the input window.
VI. File manager for opening and adding a DTASelect-filter.txt file that was obtained from the proteomic search.
VII. Button to download an example DTASelect-filter.txt file.
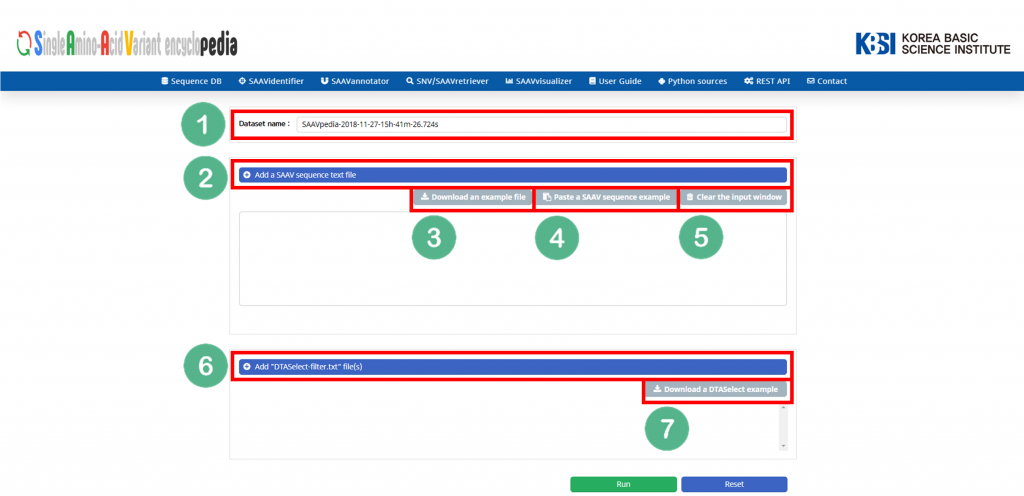
VIII. Button to export the results as SCF or EXCEL files.
IX. SAAVvisualizer modules of six kinds of plots.
X. SCF data table window.
XI. Button to go to SAAVannotator.
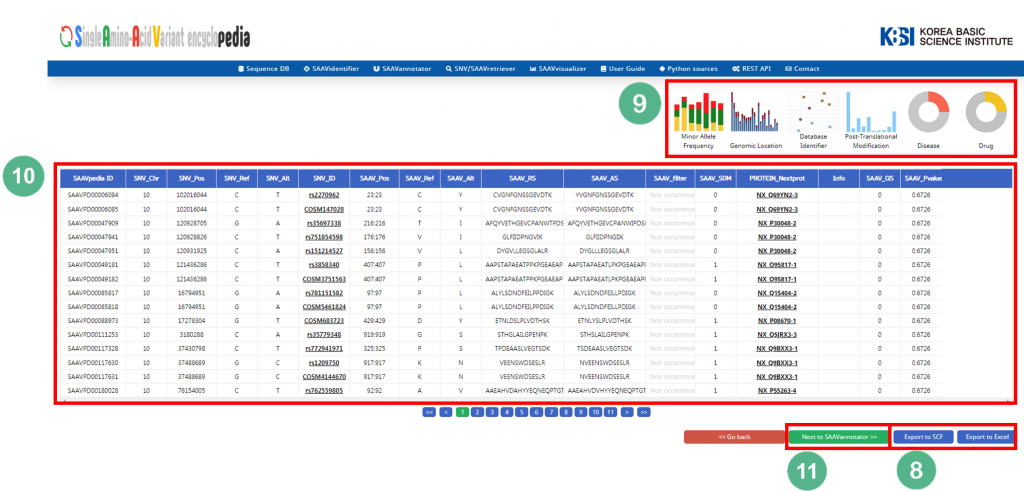
5.1.3 SAAVannotator
The SAAVannotator module provides functional annotations for comprehensive interpretation of SAAVs. It consists of a query builder, functional information database, and SAAV interpreting system.
File manager for opening and adding SCF txt files.
II. Button to download an example SCF txt file.
III. Button to automatically paste an example SCF data table.
IV. Button to clear the input window.
V. Buttons to select the annotating information of a genomic variant such as ESP MAF, 1000G MAF, and SAAV occurrence.
VI. Buttons to select the annotating information of post-translational modifications at a variant sequence.
VII. Buttons to select the annotating information of various diseases.
VIII. Buttons to select the annotating information of the identifier from genomic, transcriptomic, proteomic, pharmacological, disease, biological, or literature databases.
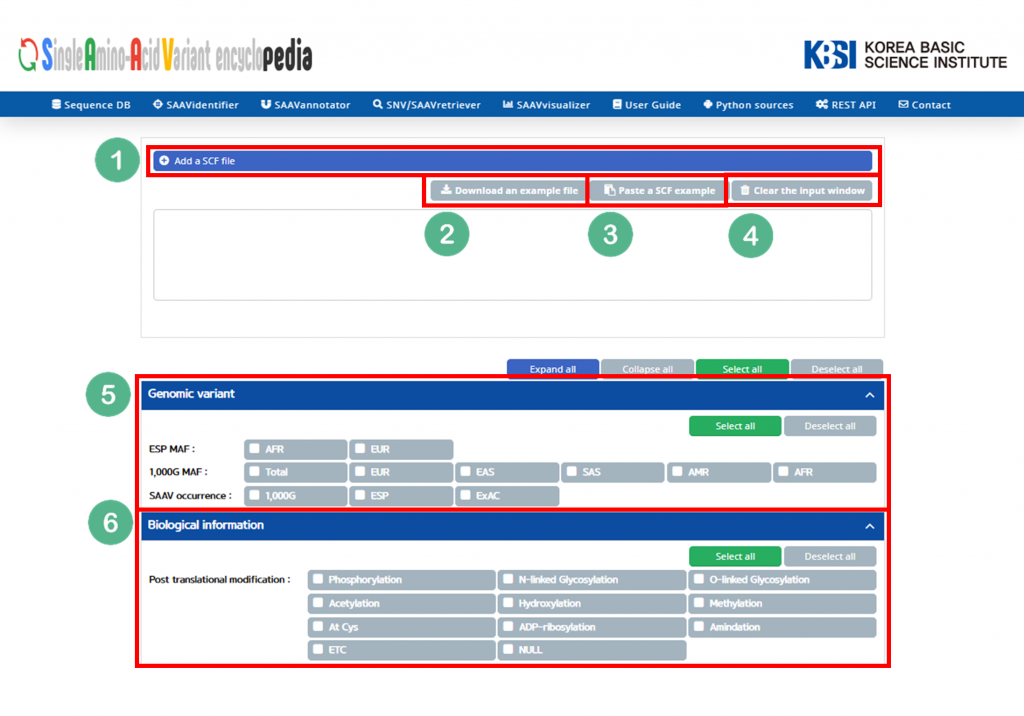
IX. Buttons to export the results in SCF or Excel file format.
X. SAAVvisualizer module of six kinds of statistical plots.
XI. SCF data table window.
5.1.4 SNV/SAAVretriever
The SNV/SAAVretriever module enables querying and bidirectional navigation between relevant conditions specific to SAAVs and nsSNVs with diverse genomic and proteomic reference data of several phenotype conditions.
I. Database Version.
II. File manager for opening and adding text files of SNV/SAAV identifiers.
III. Button to download example SNV/SAAV identifiers.
IV. Button to automatically paste example SNV/SAAV identifiers.
V. Button to clear the input window.
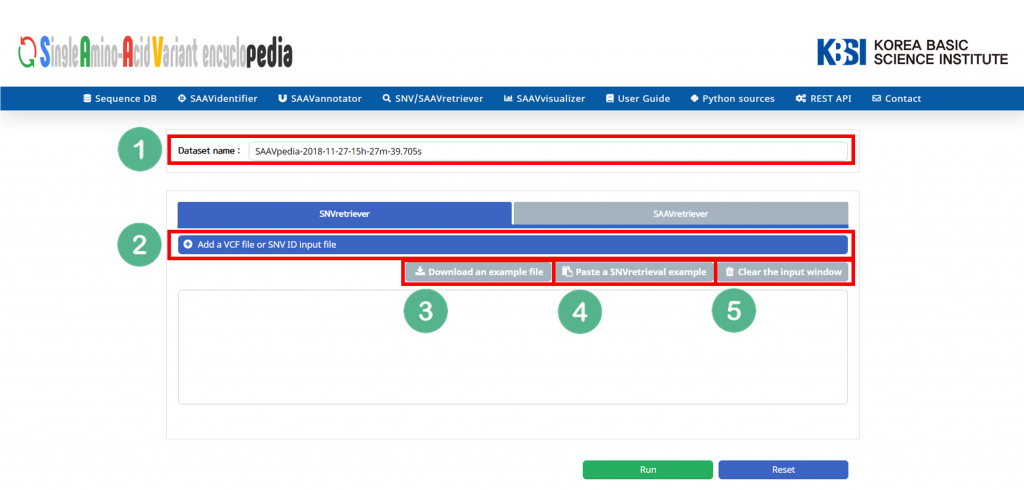
VI. Button to export the results in SCF or Excel file format.
VII. SAAVvisualizer module of six kinds of plots.
VIII. SCF data table window.
IX. Button to go to SAAVannotator.
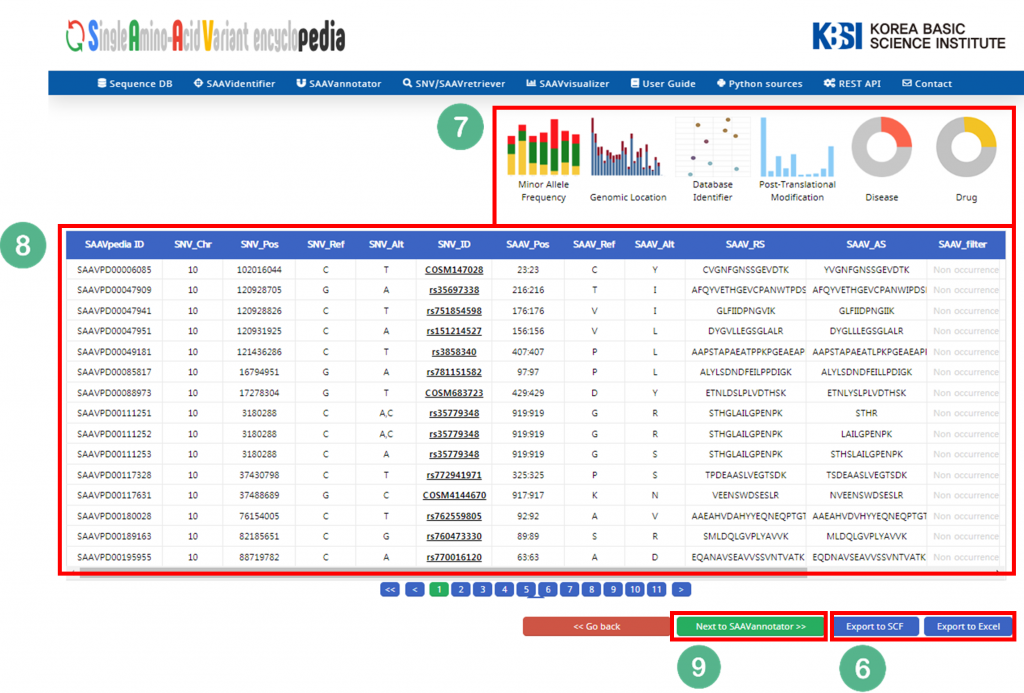
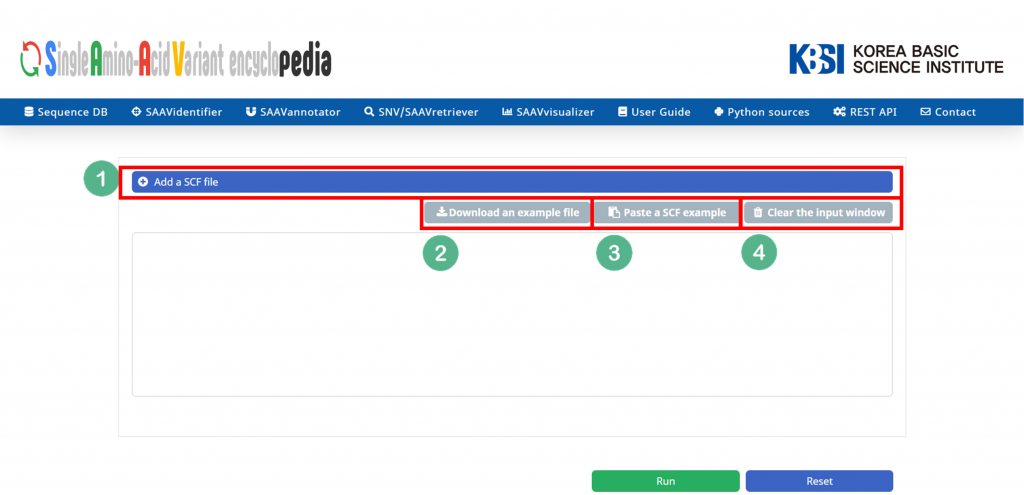
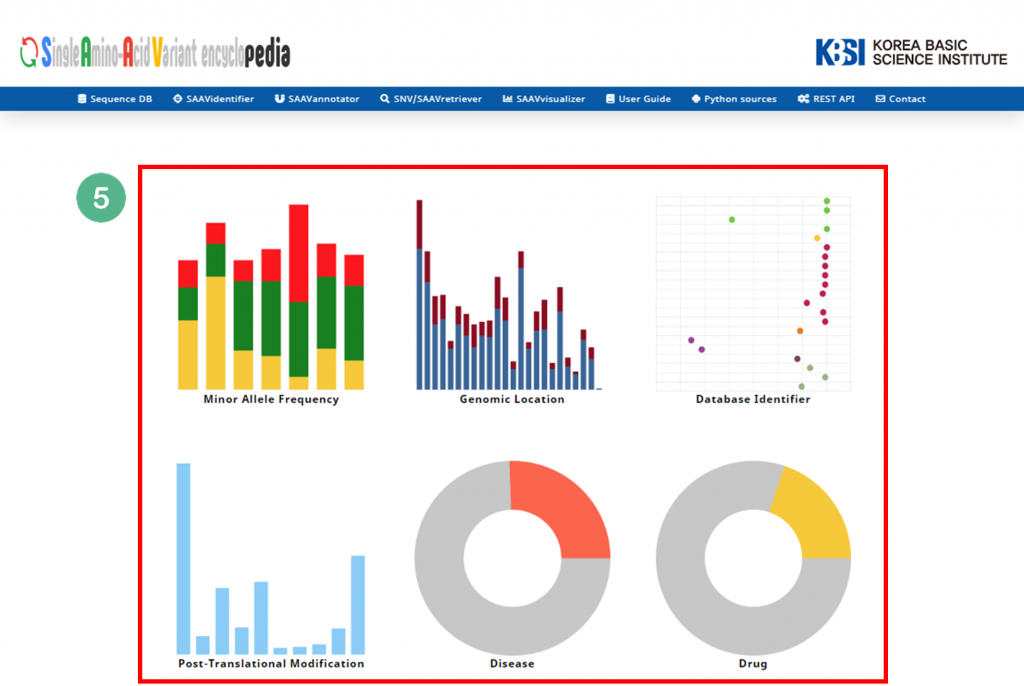
5.1.5 SAAVvisualizer
The SAAVvisualizer module can help visualize the results of the SAAVannotator and SNV/SAAVretriever modules to enable the user to efficiently grasp the meaning of the data. It provides 11 glanceable statistical summary plots based on functional elements of detected SAAVs in five categories, including minor allele frequency, genomic location, database identifier of each genomic and proteomic futures, diseases, and drugs.
I. File manager for opening and adding text files or SCF txt files.
II. Button to download an example SCF txt file.
III. Button to automatically paste example SCF txt file.
IV. Button to clear the input window.
V. Six plot icons shown in the web browser.
5.2 Python stand-alone program
5.2.1 SAAVidentifier.py
I. Open a command (or terminal) window.
II. Run SAAVidentifier module using the SAAVidentifier.input.txt file.
> python SAAVidentifier.py –input ./example/SAAVidentifier.input.txt
> more /result/SAAVidentifier*.scf

To calculate quality scores of SAAVs, the “–dta” option is used and a DTASelect file is needed (“DTASelect-filter.txt”). “SAAV_QS” and “SAAV_Pvalue” columns are added as shown below.
> python SAAVidentifier.py –input ./example/SAAVidentifier.input.txt –dta ./example/DTASelect-filter.txt
> more /result/SAAVidentifier*.scf
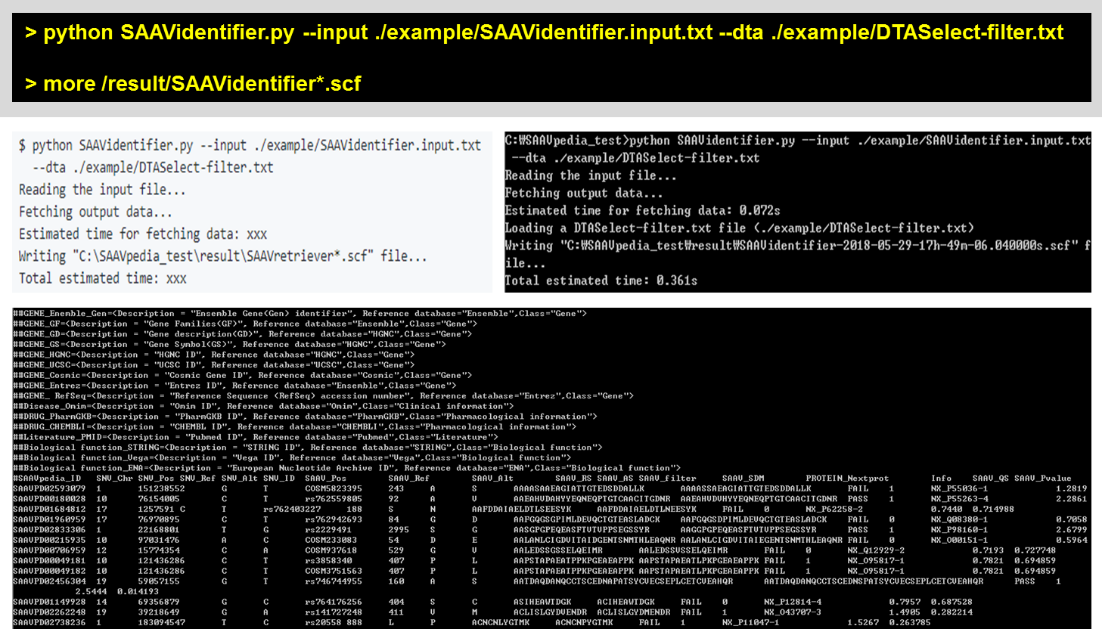
To use multiple DTASelect files for calculating quality scores, the “–dta_path” option is used. This option uses all DTAselect files that are found in not only the destination folder but also its subdirectories.
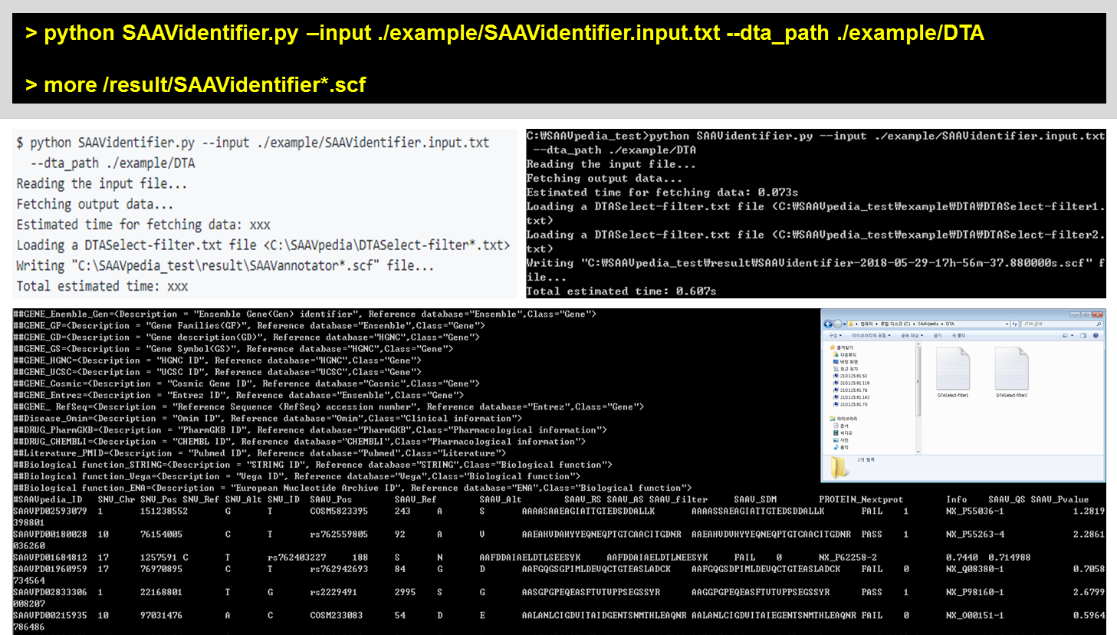
> python SAAVidentifier.py –input ./example/SAAVidentifier.input.txt –dta_path ./example/DTA
> more /result/SAAVidentifier*.scf
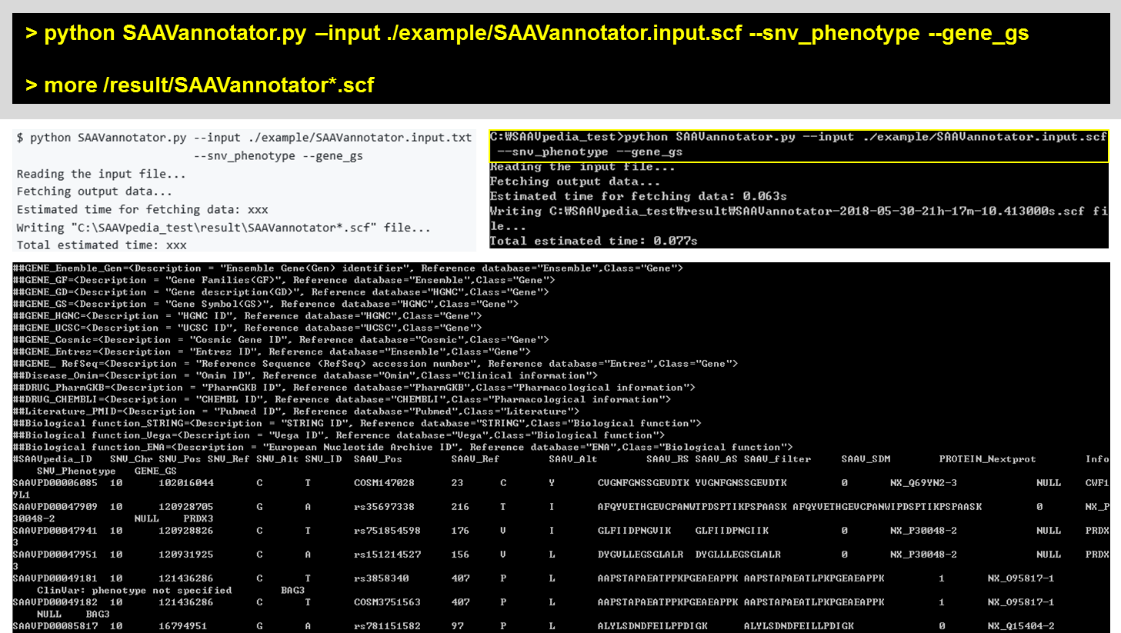
5.2.2 SAAVannotator.py
To annotate functional information to a SCF file, push the back option(s) to the SAAVannotator command. “–snv_phenotype” and “–gene_gs” (Gene Symbol) options were used in the usage example of SAAVannotator.
I. Open a command (or terminal) window.
II. Run the SAAVannotator module using the SAAVannotator.input.scf file.
> python SAAVannotatr.py –input ./example/SAAVannotator.input.txt –snv_phenotype –gene_gs
> more /result/SAAVannotator*.scf
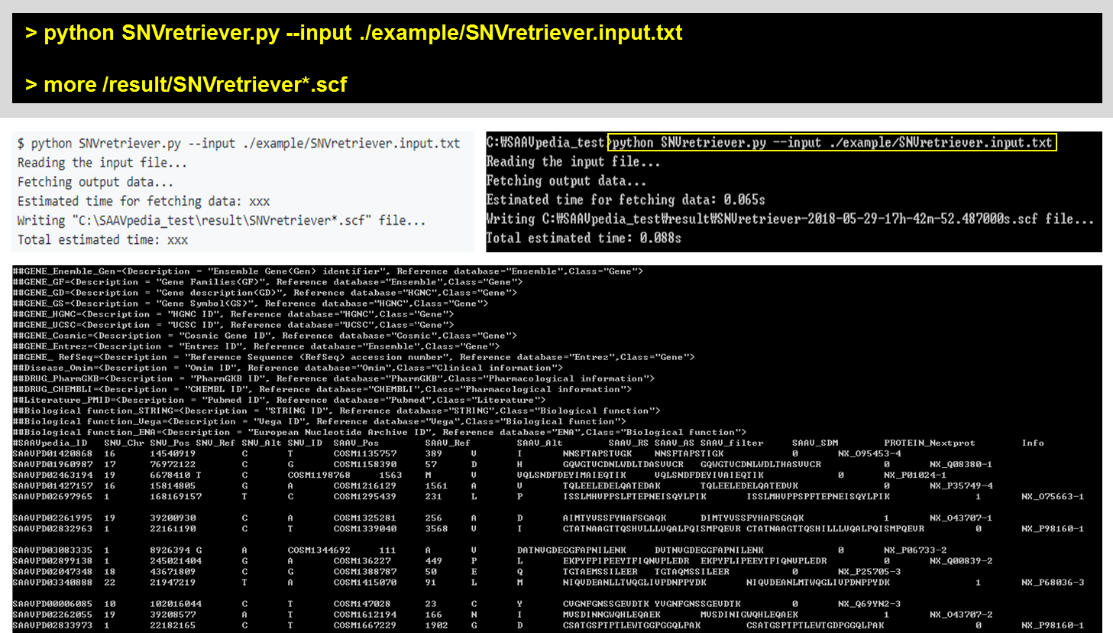
5.2.3 SNVretriever.py
The input of the SNVretriever module is a text file consisting of dbSNP and COSMIC IDs.
> python SNVretriever.py –input ./example/SNVretriever.input.txt
> more /result/SNVretriever*.scf
5.2.4 SAAVretriever.py
The input of SAAVretriever is a text file consisting of Ensembl identifiers (ENSG, ENST, and ENSP IDs), UnitProt and neXtProt accession numbers, human chromosome number, and single amino acid variant position.
> python SAAVretriever.py –input ./example/SAAVretriever.input.txt
> more /result/SAAVretriever*.scf
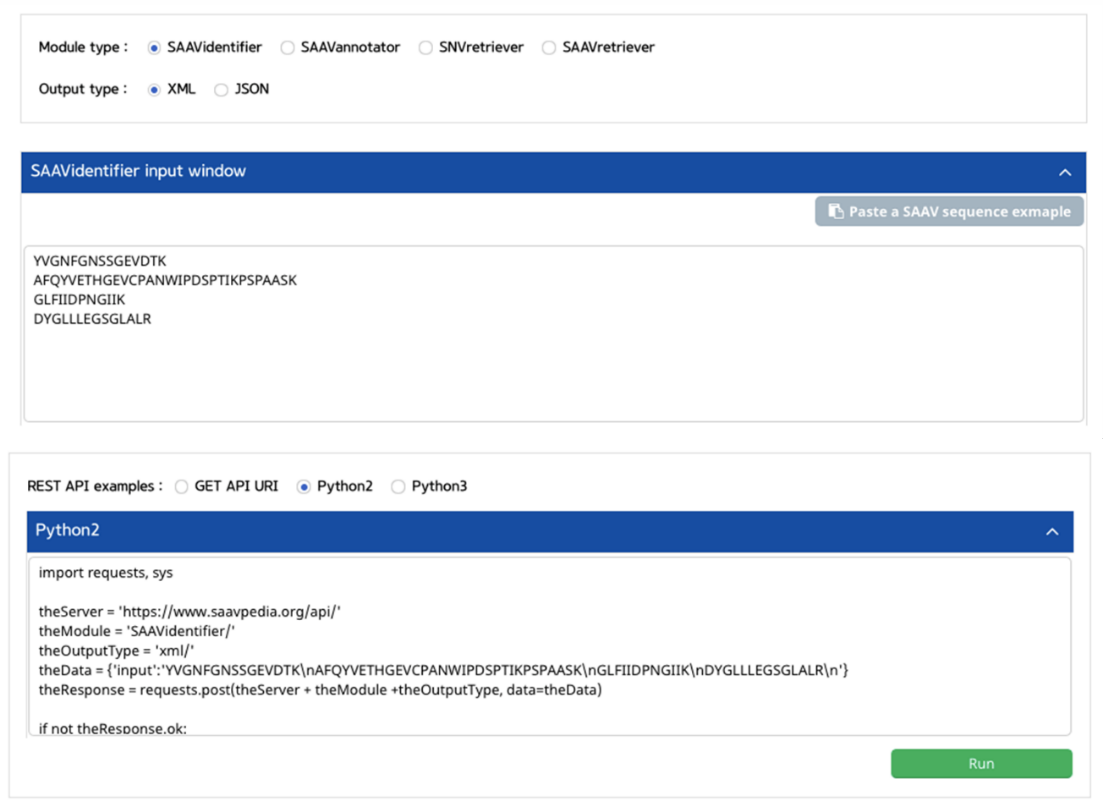
5.3 REST API
5.3.1 SAAVidentifier
I. Click “REST API” in the “Application” option of the main menu. (https://www.saavpedia.org/rest-api/)
II. Click the “SAAVidentifier” option in the module type menu.
III. Click an output type (XML or JSON).
IV. Type in SAAV peptides as the input data .
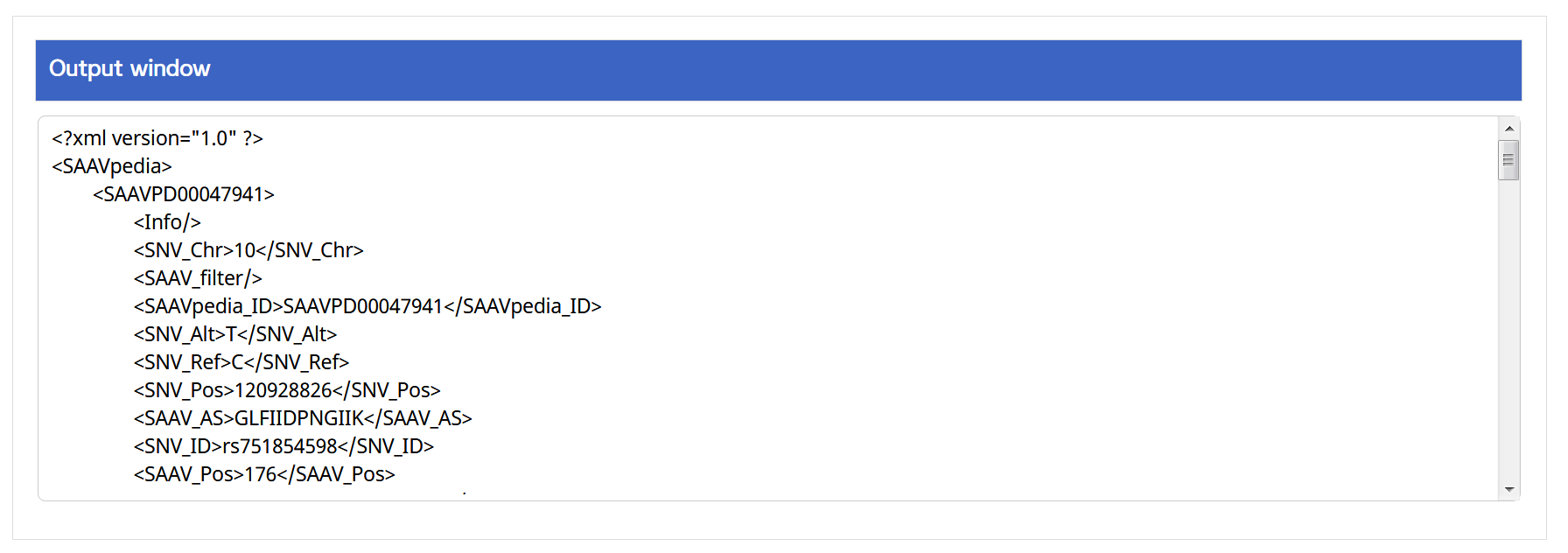
V. Click the “Run” button.
–> After running the REST Query Builder, the output window will show REST output.
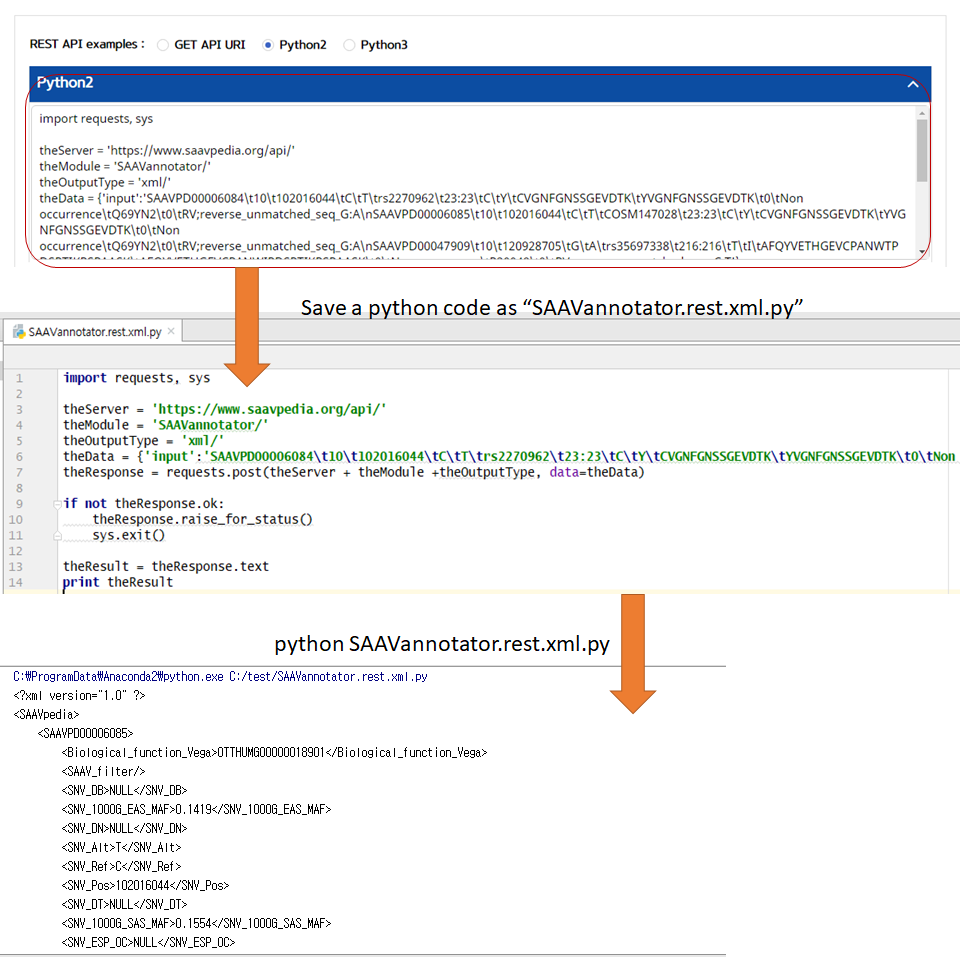
VI. Click the REST API example type.
–> After typing or pasting SAAV peptides into the input window, REST API examples are retrieved.
VII. Save the source code and run it if you want to use REST API in Python.
5.3.2 SAAVannotator
I. Click “REST API” in the “Application” option of the main menu. (https://www.saavpedia.org/rest-api/)
II. Click the “SAAVannotator” option in the module type menu.
III. Click an output type (XML or JSON).
IV. Type in the SCF file as input data.
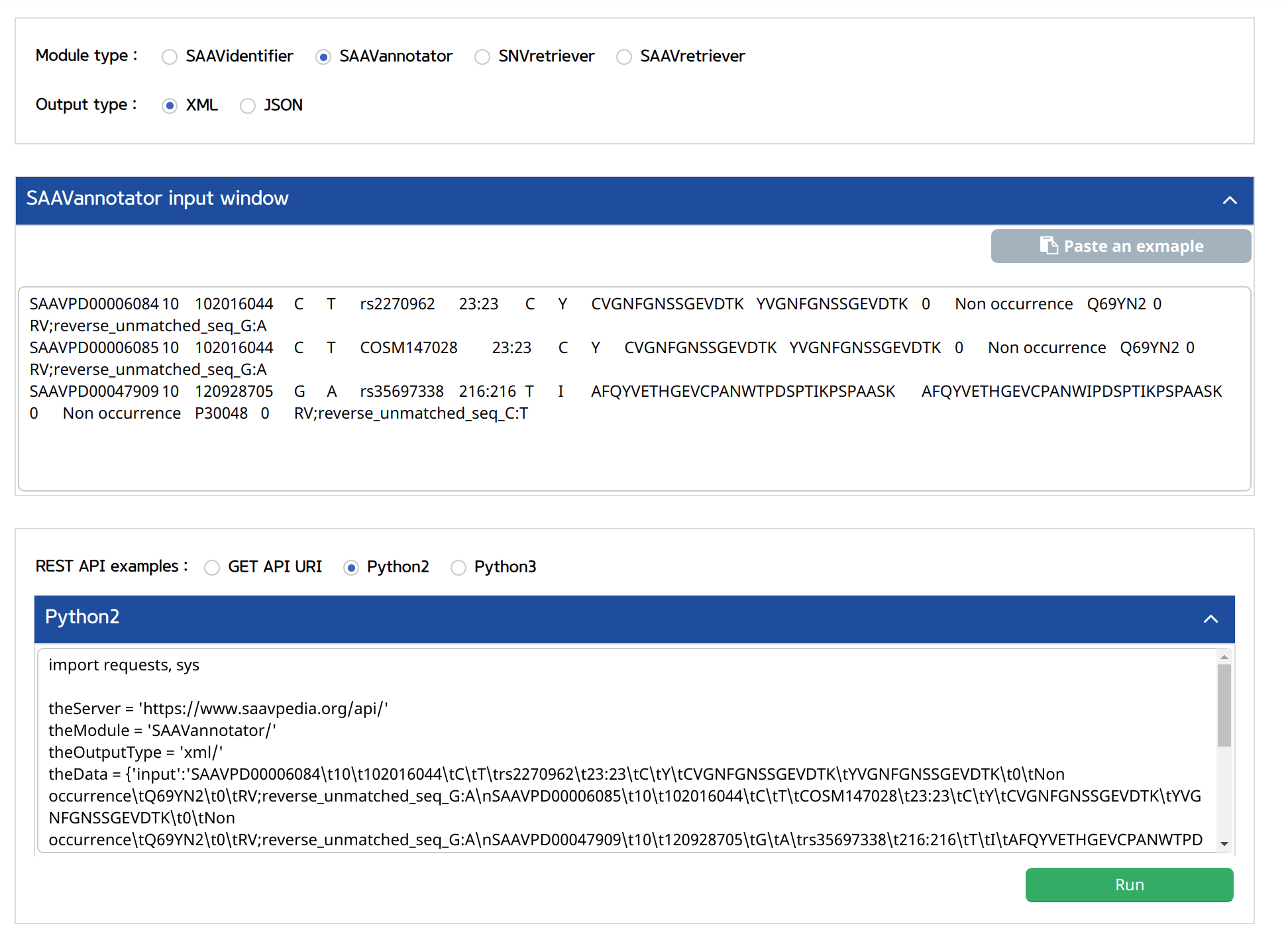
V. Click the “Run” button.
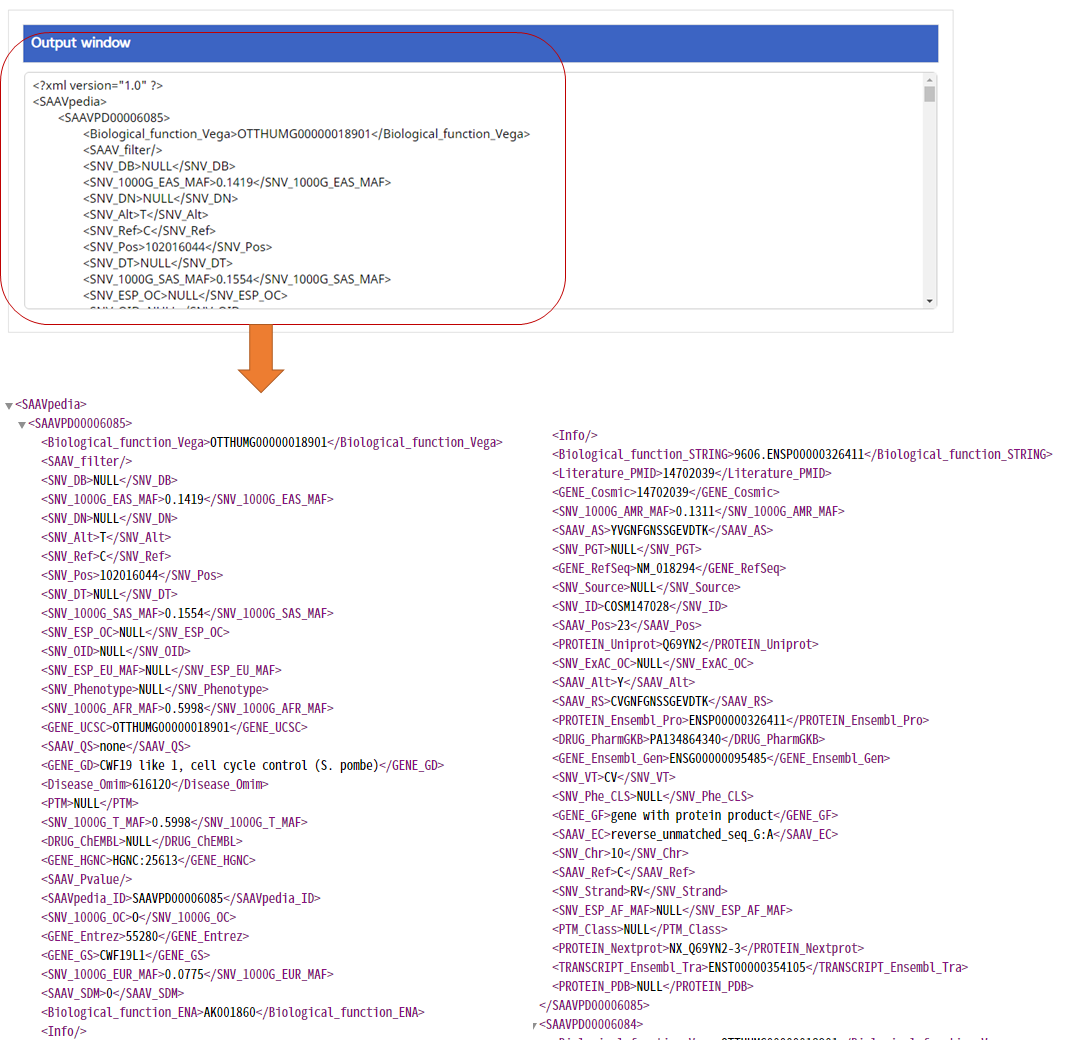
–> After running the REST Query Builder, the output window will show REST output. The REST API output of SAAVannotator is a dataset including all functional annotation terms.
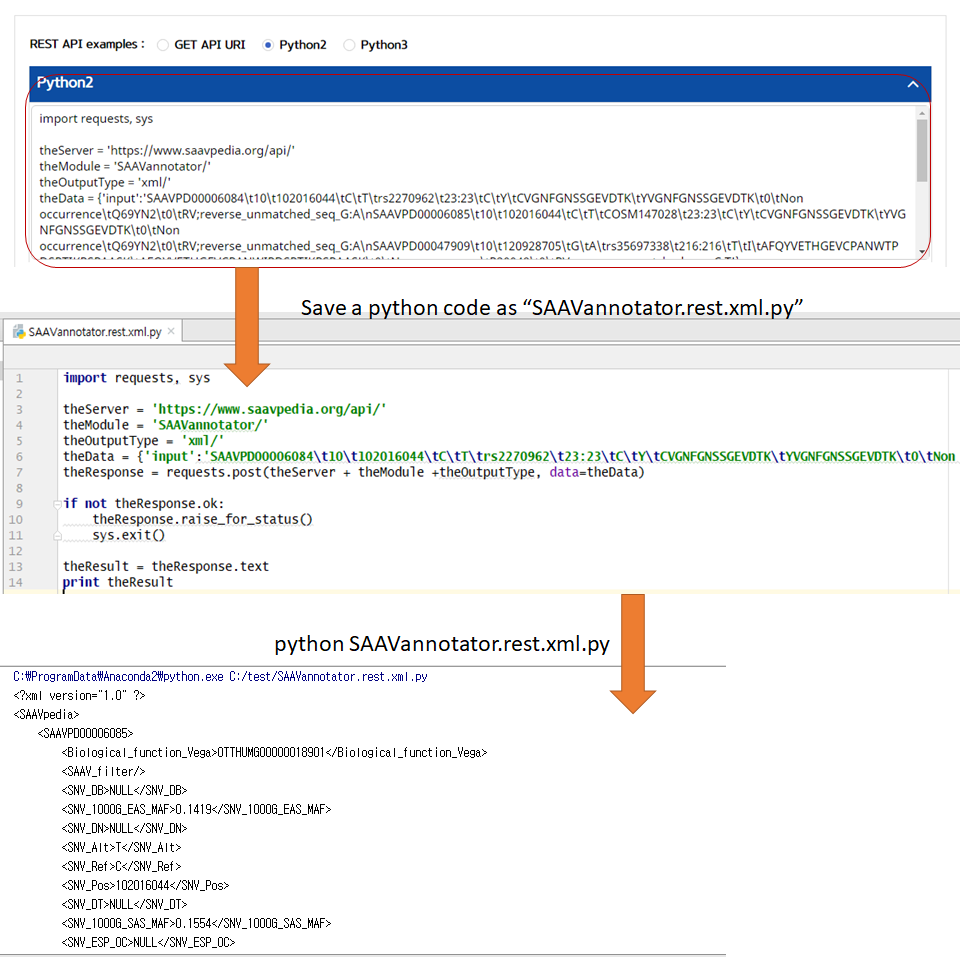
VI. Save the source code and run it if you want to use REST API in Python
5.3.3 SNVretriever
I. Click “REST API” in the “Application” option of the main menu. (https://www.saavpedia.org/rest-api/)
II. Click the “SNVretriever” option in the module type menu.
III. Click an output type (XML or JSON).
IV. Type in dbSNP or COSMIC ID as input data.
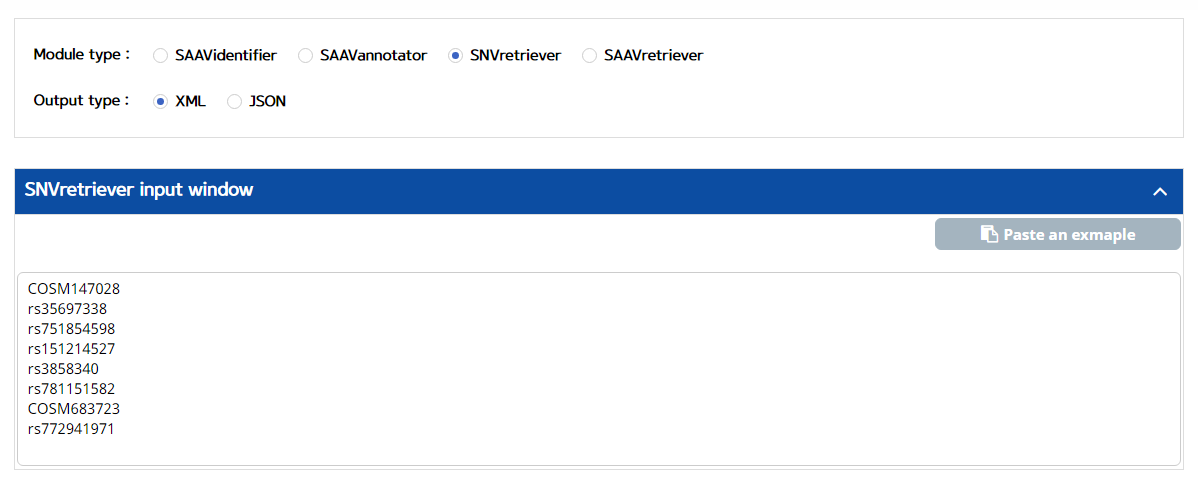
V. Click the “Run” button.
–> After running the REST Query Builder, the output window will show SNVretriever output.
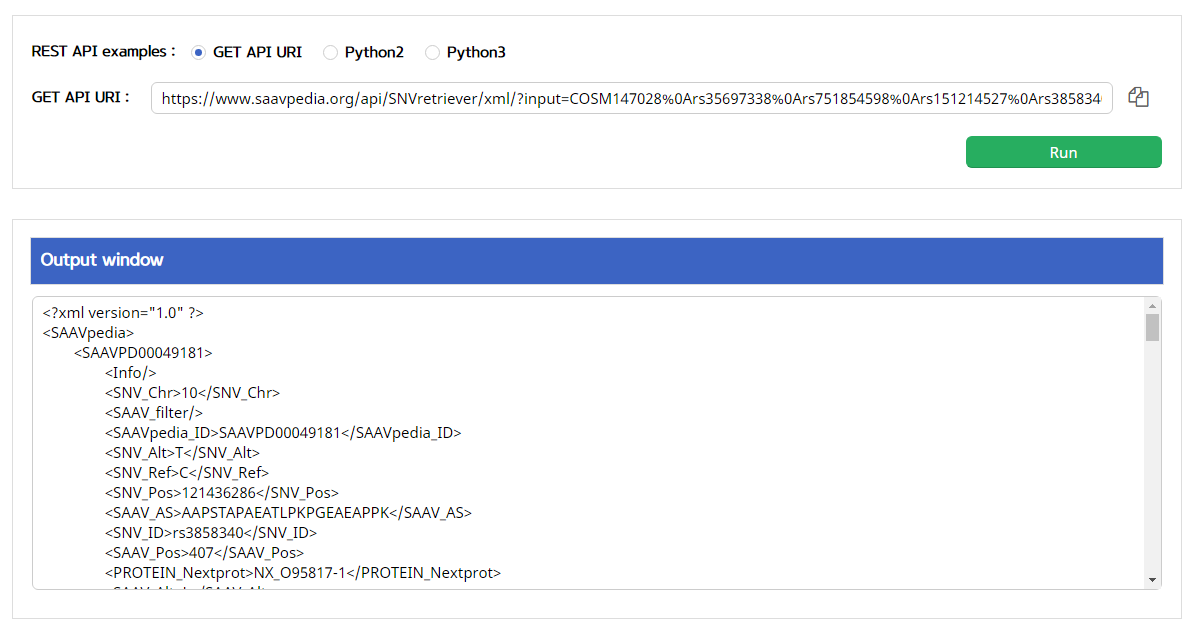
VI. Save the source code and run it if you want to use REST API in Python

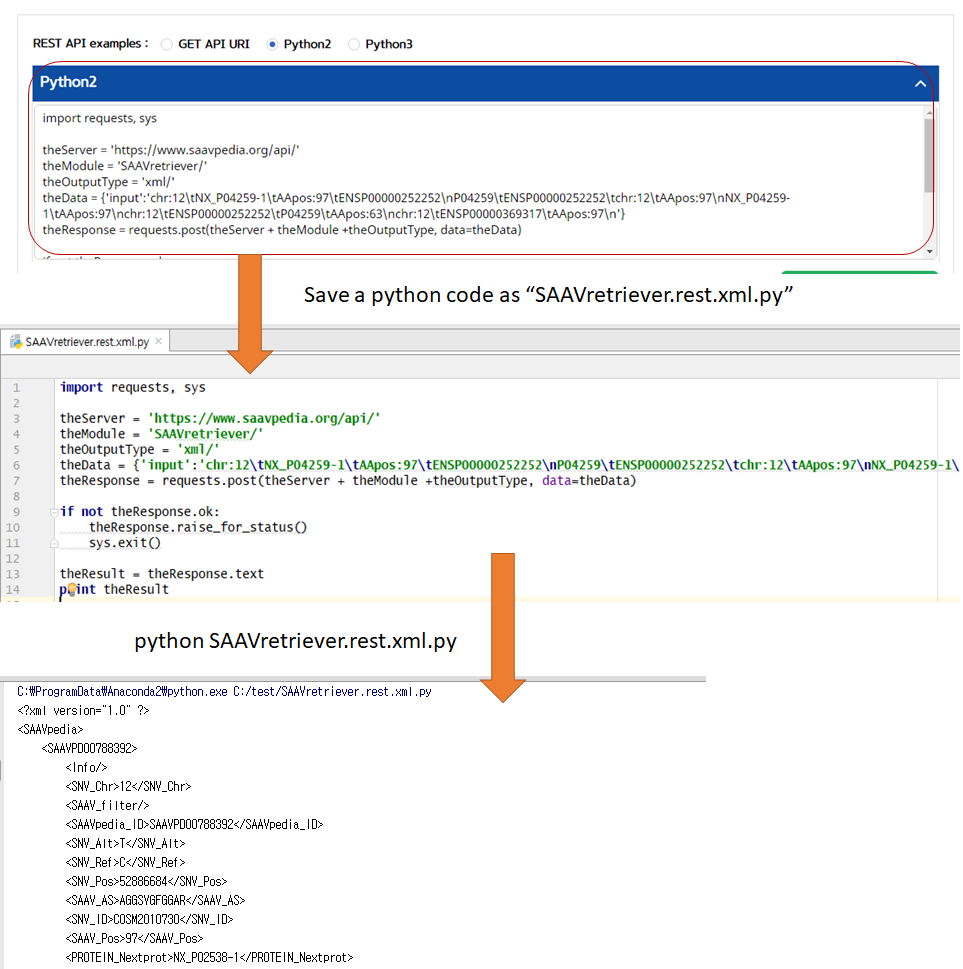
5.3.4 SAAVretriever
I. Click “REST API” in the “Application” option of the main menu. (https://www.saavpedia.org/rest-api/)
II. Click the “SAAVretriever” option in the module type menu.
III. Click an output type (XML or JSON).
IV. Type in input text consisting of Ensembl identifiers (ENSG, ENST, and ENSP IDs), UnitProt, and neXtProt accession numbers, human chromosome number, and single amino acid variant position.
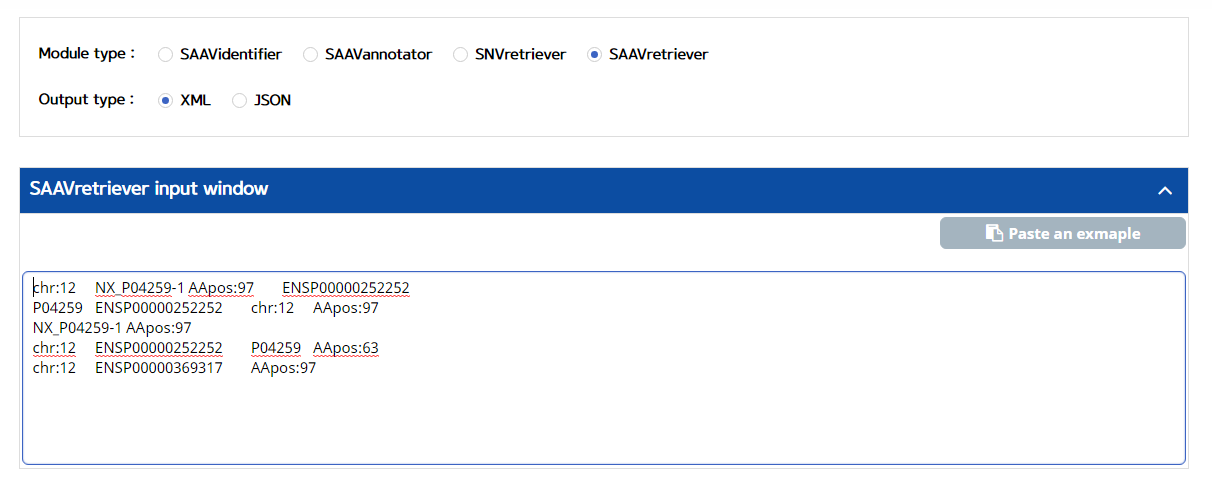
V. Click the “Run” button.
–> After running the REST Query Builder, the output window will show SAAVretriever output.
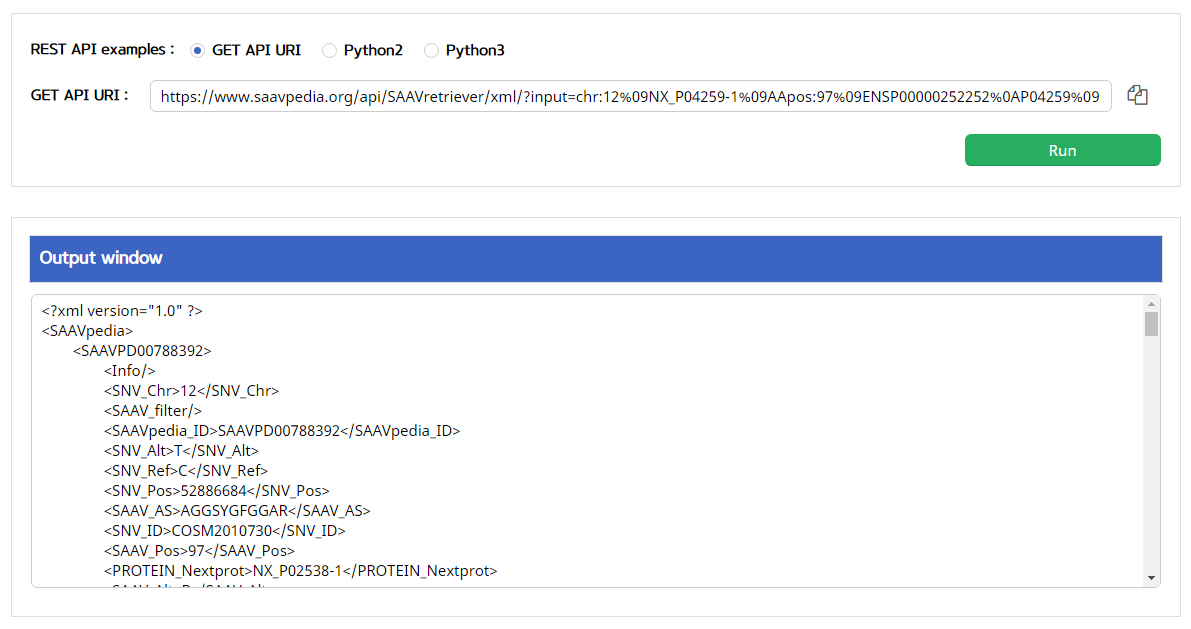
VI. Save the source code and run it if you want to use REST API in Python